相转移催化剂在雷贝拉唑钠合成中的应用
2013-11-19葛洪玉马卫兴李树安
葛洪玉, 马卫兴, 李树安
(淮海工学院 化学工程学院 江苏省海洋资源开发研究院,江苏 连云港 222005)
自1988年第一个质子泵抑制剂奥美拉唑上市以来,质子泵抑制剂已成为治疗胃酸相关性疾病的主要药物。随着对构效关系的深入研究,兰索拉唑、泮托拉唑和雷贝拉唑(1)先后在法国、德国和日本上市[1]。 1是由日本卫材公司开发的PPI类新品种,于1997年,1998年,1999年和2001年分别在日本、欧洲、美国和中国上市,其抗幽门螺杆菌的活性明显优于兰索拉唑和氧氟沙星,对耐大环内酯类抗生素的菌株也有很好的活性[2]。
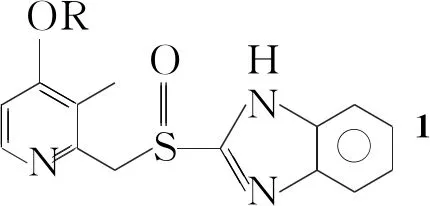
1的化学名称为2-【{[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]甲基}亚硫酰基】-1H-苯并咪唑,其结构可分为苯并咪唑环和吡啶环两个部分,合成方法主要有:(1)由取代吡啶基-甲基-硫代甲酸酯和邻苯二胺制备[3]。该方法中取代吡啶基-甲基-硫代甲酸酯制备困难,收率低;(2)由2-氯取代吡啶与苯并咪唑甲硫醚制备[4]。该方法使用丁基锂,需在低温无水条件下进行,操作困难;(3)由2-氯甲基取代吡啶与2-巯基苯并咪唑制备[5,6]。该方法原料相对易得,工艺路线较短。
本文在方法(3)合成路线的基础上加以改进:以2,3-二甲基吡啶为起始原料,经10步反应合成了1·Na(Scheme 1),其结构经1H NMR, MS和元素分析确证。在其中4步反应中引入相转移催化剂,改进了文献[5,6]方法中关键中间体较为苛刻的制备条件,提高了收率,有较好的工业应用前景。

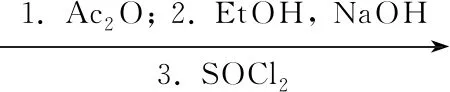
R=MeOCH2CH2CH2-
Scheme1
1 实验部分
1.1 仪器与试剂
WPS-1B型数字熔点仪(温度未校正);Bruker DRX 400型核磁共振仪(CDCl3为溶剂,TMS为内标);GCMS-QP2010 Plus型气相色谱质谱仪;LC-10AVP型高效液相色谱仪;Perkin-Elmer 2400series型元素分析仪;PSL-1000型低温合成系统。
2,3-二甲基吡啶(99%), 2-巯基苯并咪唑(98%), 3-甲氧基-1-丙醇(98%)和间氯过氧苯甲酸(85%),阿拉丁试剂;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1) 2,3-二甲基-4-硝基吡啶-N-氧化物(2)的合成
在反应瓶中加入冰乙酸180 mL,搅拌下加入2,3-二甲基吡啶32.0 g(300 mmol);滴加30%H2O240 mL,滴毕,于90 ℃反应8.0 h。加30%H2O240 mL,于90 ℃反应8.0 h。加入10%亚硫酸钠溶液100 mL,用三氯甲烷(3×100 mL)萃取,合并有机相,在低于120 ℃的油浴中旋蒸至物料重量不再减少为止。残余物为黄红色油状物,冷却得黄色固体34.0 g(也可用乙醚重结晶)。加入浓硫酸120 mL,搅拌形成悬浮液,慢慢滴加浓HNO340 mL,滴毕,缓慢升至90 ℃反应12.0 h。冷却至室温,冰水浴冷却下倒入碎冰(600 g)中,搅拌下用NaOH溶液(6.0 mol·L-1)调至pH 7.0;冷却至0 ℃,过滤,滤液用二氯甲烷(4×120 mL)萃取,合并有机相,用无水Na2SO4干燥,减压回收溶剂后用无水乙醇重结晶得黄色固体231.6 g,收率62.7%(以2,3-二甲基吡啶计),m.p.96 ℃~98 ℃(收率67.4%, 99 ℃~102 ℃[7]); Anal.calcd for C7H8N2O3: C 50.00, H 4.80, N 16.66; found C 49.87, H 4.86, N 16.58。
(2) 4-氯-2,3-二甲基吡啶-N-氧化物(3)的合成
在反应瓶中依次加入乙腈500 mL,233.6 g(200 mmol), NaCl 36.0 g(600 mmol)和浓盐酸75 mL,搅拌得淡黄色悬浮液。加入苄基三丁基氯化铵6.3 g(20 mmol),回流反应12.0 h。冷却至室温,加20%NaOH溶液140 mL,搅拌混匀后分出浅黄色有机层,残余物加水使其溶解,用二氯甲烷(3×120 mL)萃取,合并有机相,用无水Na2SO4干燥,蒸除溶剂后用丙酮重结晶得淡黄色固体3 29.5 g,收率93.9%, m.p.105 ℃~107 ℃(收率69%, 103 ℃~105 ℃[6]);1H NMRδ: 2.40(s, 3H), 2.57(s, 3H), 7.14(d,J=6.8 Hz, 1H), 8.09(d,J=6.8 Hz, 1H); EI-MSm/z: 157[M+]; Anal.calcd for C7H8NOCl: C 53.35, H 5.12, N 8.89; found C 53.31, H 5.07, N 8.81。
(3) 4-(3-甲氧基丙氧基)-2,3-二甲基吡啶-N-氧化物(4)的合成
在反应瓶中加入3-甲氧基-1-丙醇(ROH)130.0 g(1.44 mol),搅拌下加入3 31.4 g(200 mmol)和三丁基甲基溴化铵5.6 g(20 mmol); 滴加50%NaOH溶液30.0 mL,滴毕,于95 ℃~100 ℃反应10.0 h。冷却至室温,过滤,滤饼用二氯甲烷(100 mL)洗涤,滤液用二氯甲烷(4×100 mL)萃取,合并有机相,用水(2×200 mL)洗涤,无水Na2SO4干燥,减压回收二氯甲烷得棕色油状物434.8 g,收率82.5%,含量≥97%(HPLC,下同);1H NMRδ: 2.11(m, 2H), 2.20(s, 3H), 2.54(s, 3H), 3.35(s, 3H), 3.55(t, 2H), 4.10(t, 2H), 6.65(d,J=4.6 Hz, 1H), 8.16(d,J=4.6 Hz, 1H)。
(4) 2-氯甲基-4-(3-甲氧基丙氧基)-3-甲基吡啶(5)的合成
按文献[6]方法制得棕色油状物5,含量≥96%,收率62.6%,可直接用于下步反应。粗品经短硅胶柱[洗脱剂:V(石油醚) ∶V(乙酸乙酯)=1 ∶1,Rf=0.54]纯化得棕色半固状5,含量≥98.5%;1H NMRδ: 2.11(m, 2H), 2.27(s, 3H), 3.36(s, 3H), 3.56(t, 2H), 4.12(t, 2H), 4.68(s, 2H), 6.70(d,J=3.6 Hz, 1H), 8.26(d,J=3.6 Hz, 1H)。
(5) 2-【{[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]甲基}硫基】-1H-苯并咪唑(6)的合成
在反应瓶中加入甲醇溶液(甲醇250 mL+水60 mL),搅拌下加入2-巯基苯并咪唑15.0 g(100 mmol)和苄基三丁基氯化铵1.5 g(5.0 mmol),搅拌使其溶解。加入523.0 g(100 mmol),回流反应1.5 h。旋蒸除去大部分甲醇,残余物搅拌下加入饱和NaHCO3溶液(250 mL)中,于室温反应30 min。用二氯甲烷(4×100 mL)萃取,合并有机相,用水(2×200 mL)洗涤,无水Na2SO4干燥,减压回收溶剂后用乙酸乙酯重结晶得类白色固体628.2 g,收率82.2%, m.p.118 ℃~120 ℃(收率36%, 122 ℃~124 ℃[6]);1H NMRδ: 2.10(t, 2H), 2.26(s, 3H), 3.35(s, 3H), 3.56(t, 2H), 4.12(t, 2H), 4.37(s, 2H), 6.75(d,J=3.6 Hz, 1H), 7.10~7.25(m, 2H), 7.52~7.55(m, 2H), 8.33(d,J=3.6 Hz, 1H), 10.93(s, 1H); Anal.calcd for C18H21N3O2S: C 62.95, H 6.16, N 12.23; found C 62.86, H 6.08, N 12.27。
(6) 1的合成
在反应瓶中加入二氯甲烷150 mL,搅拌下加入6 20.6 g(60 mmol)和苄基三丁基氯化铵1.0 g(3 mmol),于0 ℃~5 ℃滴加KHCO3溶液(KHCO37.0 g+水30 mL),滴毕,冷却至-40 ℃,滴加间氯过氧苯甲酸溶液(间氯过氧苯甲酸12.0 g+二氯甲烷50 mL+甲醇20 mL),滴毕(约30 min),于-40 ℃反应30 min。升至室温,分液,水层用二氯甲烷(2×50 mL)萃取,合并有机相,依次用10%Na2SO3溶液(100 mL), 5%NaHCO3溶液(100 mL),水(2×200 mL)和饱和食盐水(100 mL)洗涤,无水Na2SO4干燥,减压回收溶剂后加入丙酮(50 mL)中,搅拌均匀后置冰箱中冷冻过夜。过滤,真空干燥得白色固体111.8 g,收率54.8%, m.p.98 ℃~100 ℃(收率82%, 99 ℃~101 ℃[8]);1H NMRδ: 1.93~2.06(m, 2H), 2.12(s, 3H), 3.35(s, 3H), 3.52(t, 2H), 4.12(t, 2H), 4.69(d,J=7.6 Hz, 2H), 4.85(d,J=7.6 Hz, 1H), 6.83(d,J=3.6 Hz, 1H), 7.07~7.28(m, 2H), 7.36~7.60(m, 2H), 8.27(d,J=3.6 Hz, 1H); Anal.calcd for C18H21N3O3S: C 60.15, H 5.89, N 11.69; found C 60.21, H 5.86, N 11.60。
(7)1·Na的合成
在反应瓶中依次加入无水甲醇100 mL和111.0 g(30 mmol),搅拌下滴加甲醇钠/甲醇溶液[钠0.7 g(30 mmol)+甲醇15 mL],滴毕,于室温反应1.0 h。过滤,蒸除大部分溶剂得黏稠油状物。搅拌下加入无水乙醚100 mL,置冰箱中冷冻析晶,3 d后取出过滤,滤饼用少许冷无水乙醚洗涤,真空干燥得白色固体1·Na 8.9 g,收率77.8%,含量≥99.0%, m.p.138 ℃~140 ℃(分解)[收率87%, 140 ℃~141 ℃(分解)[5]];1H NMR(DMSO-d6)δ: 2.01(t, 2H), 2.19(s, 3H), 3.26(s, 3H), 3.49(t, 2H), 4.10(t, 2H), 4.56(m, 2H), 6.81~6.93(m, 3H), 7.36~7.58(m, 2H), 8.27(d,J=3.6 Hz, 1H); Anal.calcd for C18H20N3O3SNa: C 56.68, H 5.29, N 11.02; found C 56.58, H 5.33, N 11.08; EI-MSm/z: 382[M+1]。
2 结果与讨论
2.1 相转移催化剂在合成中的应用
Tascioglu S[9]就表面活性剂在有机溶剂/水体系中的作用及由此带来的诸多不同性质做了详细的叙述,加入相转移催化剂可以改变反应组分的解离电位和氧化还原性质以及物理性质,从而可以改变反应的活性,最终导致反应体系的局部浓度加大,由此提高反应的选择性和效率,使反应更单一,更完全,纯度更高。
在1的全合成中,文献[8,10]方法也使用相转移催化剂,但仅仅局限于在一步或两步反应中。实验中我们发现,在中间体3,4,6和1的合成中,反应体系都是有机溶剂/水体系,受文献启发,考虑使用相转移催化剂来优化反应。通过试验,发现在这几步反应中使用相转移催化剂均能加快反应速度,使反应更完全,同时反应效率和选择性好,后处理比较简单,克服了其它方法重现性差,产率和产物纯度不稳定的缺点。在1的合成中,同时在4步反应中使用相转移催化剂还未见文献报道。
2.2 1在无水体系中成盐和结晶
苯并咪唑型质子泵抑制剂在酸性或中性条件下易于降解,制备成碱性盐的形式可以增强其稳定性。1的成盐,文献报道的方法大都以1与氢氧化钠水溶液反应,反应体系中含有水,同时反应过程中还生成部分水。1·Na在水中有一定的溶解度,因此给产品的脱水和结晶带来难度。本文将1的成盐和结晶都在无水体系中进行,反应过程中也没有水的生成,产品结晶稳定,不易吸潮变质;所得晶体晶型较好,易过滤及与溶剂有效分离;所用溶剂毒性小,易于除去,易实现工业化。
2.3 2的合成
在2的合成中,首先要制备2,3-二甲基吡啶-N-氧化物,不经过纯化而直接用于后面的硝化反应。为了硝化反应能够很好地进行,2,3-二甲基吡啶-N-氧化物的制备中杂质含量要控制好,氧化反应结束后蒸除溶剂和水时,油浴的温度不能超过120 ℃,否则料液的颜色会加深,对下步反应中产物的收率和色泽带来影响。残余溶剂和水一定要蒸干净,最终得到黄红色油状物稍冷却即能固化成黄色固体,如果溶剂和水没有蒸干净,黄红色油状物不能固化或固化不完全,对后面的硝化反应影响较大,甚至不能进行反应。
[1] 孙忠实. 质子泵抑制剂的新突破——雷贝拉唑[J].中国药学杂志,2003,38(4):307-309.
[2] 高国芳,沈利君,俞静玉. 雷贝拉唑的药效学与药功学特点[J].医药导报,2004,23(6):401-402.
[3] 程卵生,王庆河,潘莉,等. 吡啶衍生物的制备与方法[P].CN 1 102 411A,1995.
[4] Fischli A, Krasso A, Szente A,etal. (Bezimidazol-2-yl)-pyridinium compounds[P].US 4 766 133,1988.
[5] Souda S, Ueda N, Miyazawa S,etal. Pyridine derivatives having anti-ulcerative activity[P].US 5 045 552,1991.
[6] 徐宝财,刘家胜,周雅文,等. (R)-2-{[4-(甲氧基丙氧基)-3-甲基吡啶-2-基]甲基亚硫酰基}-1H-苯并咪唑的合成[J].有机化学,2008,28(12):2155-2158.
[7] Fischli A, Krasso A, Ramaz H,etal. (Bezimidazol-2-yl)-pyridinium compounds[P].US 4 634 710,1988.
[8] 邱飞,刁勇. 雷贝拉唑的合成[J].中国医药工业杂志,2010,41(1):9-10.
[9] Tascioglu S. Micellar solutions as reaction media[J].Tetrahedron,1996,52(34):11113-11152.
[10] 蒋军荣,刘学峰,陈晓芳. 2-氯甲基-3-甲基-4-[(3-甲氧基)丙氧基]-吡啶盐酸盐的合成[J].合成化学,2007,15(3):391-393.
