蛋白质功能预测方法概述
2013-11-14沈素萍方慧生陈凯先
刘 言,沈素萍,方慧生*,陈凯先
(1.中国药科大学生命科学与技术学院,江苏南京210009;2.上海药物研究所药物发现与设计中心,上海201203)
1 引言
基因组学和蛋白质组学在过去十年的发展过程中产生了大规模的新的蛋白质序列和试验数据,科学家为了确定这些新序列的功能借助计算机手段进行了大量的研究[1-2]。在过去的二十年里,人们利用计算机技术对蛋白质功能进行预测的文章发表了上 千 篇 之 多 (http://www.ncbi.nlm.nih.gov/pubmed),大部分是基于序列相似性、基于结构域、基于相互作用网络等方法预测,再利用生物学知识来进行解析。本文综合阐述了迄今为止蛋白质功能预测的分类,大致可分为四类:(1)基于序列相似性预测方法;(2)基于蛋白质相互作用网络预测方法;(3)基于结构相似性预测方法;(4)其他预测方法。
2 蛋白质功能
蛋白质功能对于客观环境很敏感:给定的发挥作用的空间环境不同、规定的作用时间不同都可以使蛋白质所表现出来的功能是有差异性的。为了使功能预测的结果更加准确,Bork等提出了一种蛋白质功能类型的分类[3],按蛋白质发挥作用的平台不同将蛋白质功能分为分子功能,细胞功能和生理功能。很明显,这三个类型不是独立存在的,而是如图2那样等级相关的。现如今在蛋白质功能预测中最常用的是GO分类,Gene Ontology分类从细胞组成、分子功能和生物学途径三方面描述蛋白质的性质与功能。分子功能是描述其分子生物学活性,如催化活性、结合活性,可以具体到腺苷酸环化酶活性或钟形受体结合活性等;生物学途径是细胞生长和维持、信号转导过程,更狭义可描述为在嘧啶代谢或α-配糖基的运输等具体过程。所以蛋白质功能预测的最终想得到结果是:这个新序列在细胞中充当什么组分,在哪个生物学过程中起作用,起着什么样的作用。
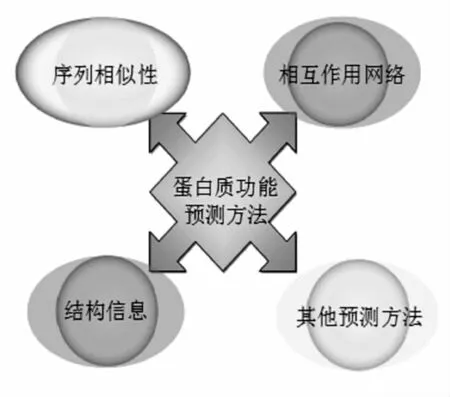
图1 蛋白质功能预测方法的分类Fig.1 Protein function prediction methods
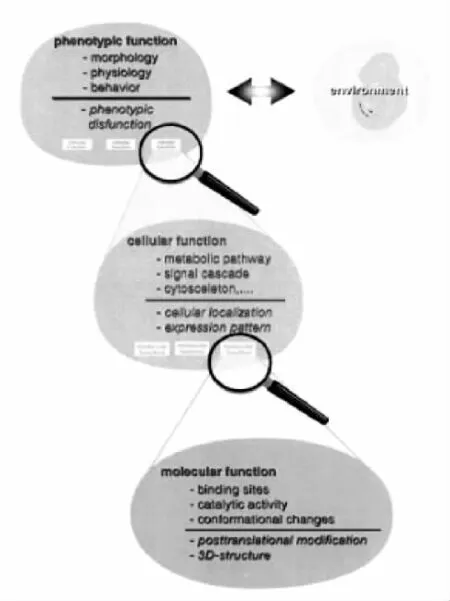
图2 蛋白质功能类型分类Fig.2 Protein function types classification
3 蛋白质功能预测的方法
蛋白质功能预测方法可粗略分为基于序列相似性预测、基于蛋白质相互作用网络预测、基于结构相似性预测和其它不依赖于相似性的预测方法。我们将分别列举近年来基于这四类方法所做的蛋白质功能预测,以及它们各自的优势与弱势。
3.1 基于序列相似性预测蛋白质功能
基于序列相似性是较早的一种功能预测的方法,它是基于序列相似,功能相似的假说建立的。最传统的方法是对新序列进行BLAST或PSI-BLAST搜索[4],通过产生的E值选择与新序列高度相似的序列(一般序列一致性要在40%以上[5]),由已知序列功能推断出新序列的功能。但随着研究的不断深入,这种方法被证明是不可靠的[6],因为序列同源性不等于功能一致性[7]。基于序列同源性的模型的建立过于依赖蛋白质之间的相似程度,所以只能适用于与功能已知蛋白质有很高同源性的新蛋白序列的功能预测。并且随着同源性降低,建立模型的误差增加。
Hawkins[8-9]分别通过提取 Go terms 和对 Go terms评分的方法对传统的PSI-BLAST搜索进行拓展,包括从亲缘关系较远的序列进行注释、应用新的数据挖掘工具、功能相关矩阵、得分密切相关的注释对,开发出可以通过降低分辨率来增加功能注释的普及型的方法PFP(protein function prediction)。PFP方法综合考虑了GO terms评分和GO terms与其亲代GO terms之间的功能相关性。从而不需要精准的匹配模式或蛋白质结构信息,只需要较弱相似序列就可以推断出新序列的功能,结果的精确度和覆盖范围比传统的PSI-BLAST结果高出五倍不止。由 AFP - SIG 05[10]和 CASP7[11]两个高级别的比赛结果就可以证明PFP方法是很成功的。
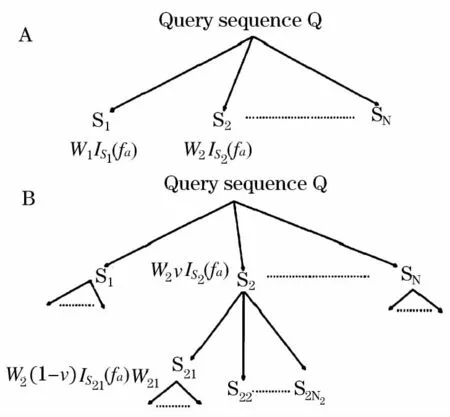
图3 ESG方法建立的序列相似图谱Fig.3 The sequence similarity map establish by ESG method
Chitale[12]于09 年建立了 ESG(extended similarity group)方法,此方法执行迭代序列数据库搜索并且对新序列进行GO terms注释。注释就是给每条序列制定一个概率,这个概率是基于蛋白质序列相似图谱(图3)中multiple-level neighbors的亲缘相似评分所得的。图4中用funsim(Fundamental Simulation Instruction Method)对 PFP、Top-PSIBLAST、ESG三种方法进行了对比,从图中可以看出ESG方法所产生结果较好。
图3 PSI-BLAST搜索得到的序列相似图谱,序列Q经过PSI-BLAST搜索返回N条序列,称为ESG first level,对 ESG first level进行 PSI- BLAST再返回N条序列称为ESG second level,以此类推得到ESG multiple-level,各序列之间称为multiplelevel neighbors。

图4 根据funsim打分得到的结果准确度对比Fig.4 Results accuracy compared get though funsim scoring
基于序列的蛋白质功能预测考虑的是独立的蛋白质序列,未考虑蛋白质之间的相互作用。而蛋白质是通过与其它蛋白质直接或间接相互作用而执行功能的。所以要从序列预测蛋白质的功能应该将与其相互作用的蛋白质序列一同考虑在内。
3.2 基于相互作用网络预测蛋白质功能
基于PPI(protein-protein interaction)的预测方法主要用于从多个蛋白质序列中寻找有相互作用和关联进化的蛋白质或从PPI数据库中提取信息,预测效果依赖于基因组数目和PPI数据库的准确程度。由 Bader等[13]开发的 Pathguide(http://www.pathguide.org)提供大部分PPI相关的数据库列表和链接,表1列出了部分PPI数据库。根据这些数据库中提取的蛋白质相互作用数据,人们可以构建相应的相互作用网络。在相互作用网络中,一般用节点(node)来表示蛋白质,而连接两个节点的边(edge)表示蛋白质之间是否存在相互作用关系。

表1 蛋白质相互作用数据库Table 1 Protein interaction database
目前,利用相互作用网络进行功能注释主要有 两种方法,即直接注释方法(direct annotation schemes)[14-16]和基于模块的方法(module - assisted schemes)[17-18]。
3.2.1 直接注释方法
为了有效的打击商业贿赂犯罪,许多国家都采取一系列的措施来建立和完善举报人保护和奖励制度:比如德国规定,不公开举报人和证人的住址,并且允许通过匿名方式进行举报。同时德国还对揭露商业贿赂的信息来源予以法律保护,任何人都不能对信息的来源进行调查[12]。再比如日本对举报人的保护制度制定专门的《公益举报人保护法》,保护那些揭露公司主管或人员徇私舞弊的举报人。该法规定要对举报人的身份予以保密,不得以任何形式泄露举报人。对举报商业贿赂的单位和个人,给予重奖。公司不得以任何理由解雇或用其他任何形式打击报复举报人。如果发生此类情况,将按有关法律严肃处理[13]。
Vazquez[14]等首先采用基于分割的方法(cutbased approaches)将图论法引入蛋白质功能注释研究中。其基本思路是:对一个未知功能蛋白质赋予某种功能,要使得注释为相同功能的蛋白质(未注释或者已注释)的连接数目最多。Hu[15]综合考虑了PPI信息和序列的生物化学/物理化学特征,当未注释蛋白质与已知功能的蛋白质几乎没有序列相似性时,也可以获得相关的PPI信息。并应用此方法对鼠源蛋白质功能进行预测,在训练集合测试集中一阶成功率分别为69.1%和70.2%。构建蛋白质相互作用网络时通常是从注释蛋白质到非注释蛋白质做一个单向的预测。而真正的生物学过程中蛋白质是有流动性的,它们之间有动态的相互作用,从而产生了一个外环境稳定但内部千变万化的框架。Chi[16]首次将蛋白质之间动态相互作用加入到了预测过程中,方法是先给未注释的蛋白质指派一个最初的功能,然后计算此蛋白质和与其相邻的蛋白质之间的最初相似性。用基于KNN的预测算法为未注释的蛋白质预测一个新的功能,用这个新预测的功能代替最初的功能,再重新计算该蛋白质和与其相邻的蛋白质之间的相似性,在进行下一轮的计算。直到未注释的蛋白质和与其相邻的蛋白质之间的相似性达到一个稳态平衡时结束。正确定义蛋白质之间的相似性迭代法比非迭代法显示了更好的准确度和召回率,同时可行性和有效性也得到了提高。
3.2.2 基于模块预测方法
Rives[17]等人就提出一个假设,认为同一个模块中的蛋白质成员更加可能拥有最短的路径距离谱(path distance profiles)。根据这个假设,所有短路径的蛋白质对聚成一类。这个方法实施比较复杂,很难在整个基因组水平上的网络上进行分析,但在一些子网络中它已经得到很好的应用,比如对酿酒酵母的核蛋白的相互作用网络分析。Janusz[18]整合了发育和癌症研究项目的基因表达谱和蛋白质相互作用图谱提供了一个有系统和全局代表性的组合网络模块。并开发了一种新方法 Network-Guided Forests,该方法是以间接网络域相关的决策树来确定网络模块的生物或临床结果,由此产生的网络签名证明在不同样本队列之间的稳健性和捕捉发展与疾病的因果关系。
3.3 基于结构信息预测
最早基于结构进行蛋白功能注释的方法是找到一个结构相似的蛋白,将其功能转移给前一个蛋白,如在蛋白序列中的情况一样。然而这种方法并不能够单独被用来预测蛋白质功能,因为它的准确性只有20% -50%[19],结果是不足以令人采纳的。所以从3D结构衍生了多种其他的可能预测蛋白质功能的方法(如图5)。
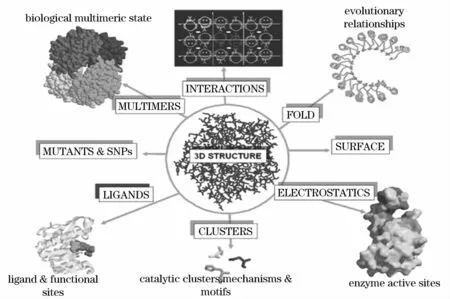
图5 3D结构衍生出的多种可能的功能预测的方法[20]Fig.5 A variety of possible functions of prediction methods derived from 3D structure
结构基序是存在于几个相关蛋白质结构中的一个蛋白的三维亚结构,它与功能息息相关。最为大家所熟知的结构基序是在许多DNA结合蛋白中均能找到的螺旋-转角-螺旋(HTH)基序。Leo C等[21]对人类 TRIM 家族中 TRIM20(pyrin)和TRIM21两个与疾病相关的蛋白进行了研究,阐明了C末端PRYSPRY区域是如何影响TRIM的功能。鉴于大部分蛋白质功能研究都是针对特异性蛋白这一状况,Akira R[22]提取了PDB数据库中所有蛋白质结构,然后从中提取出所有的结合位点,通过多次聚类得到复合基序(如图6),将复合基序分组,根据各组的复合基序的功能特征来确定蛋白质的功能。这一方法的不局限性是蛋白质功能预测的一大突破。
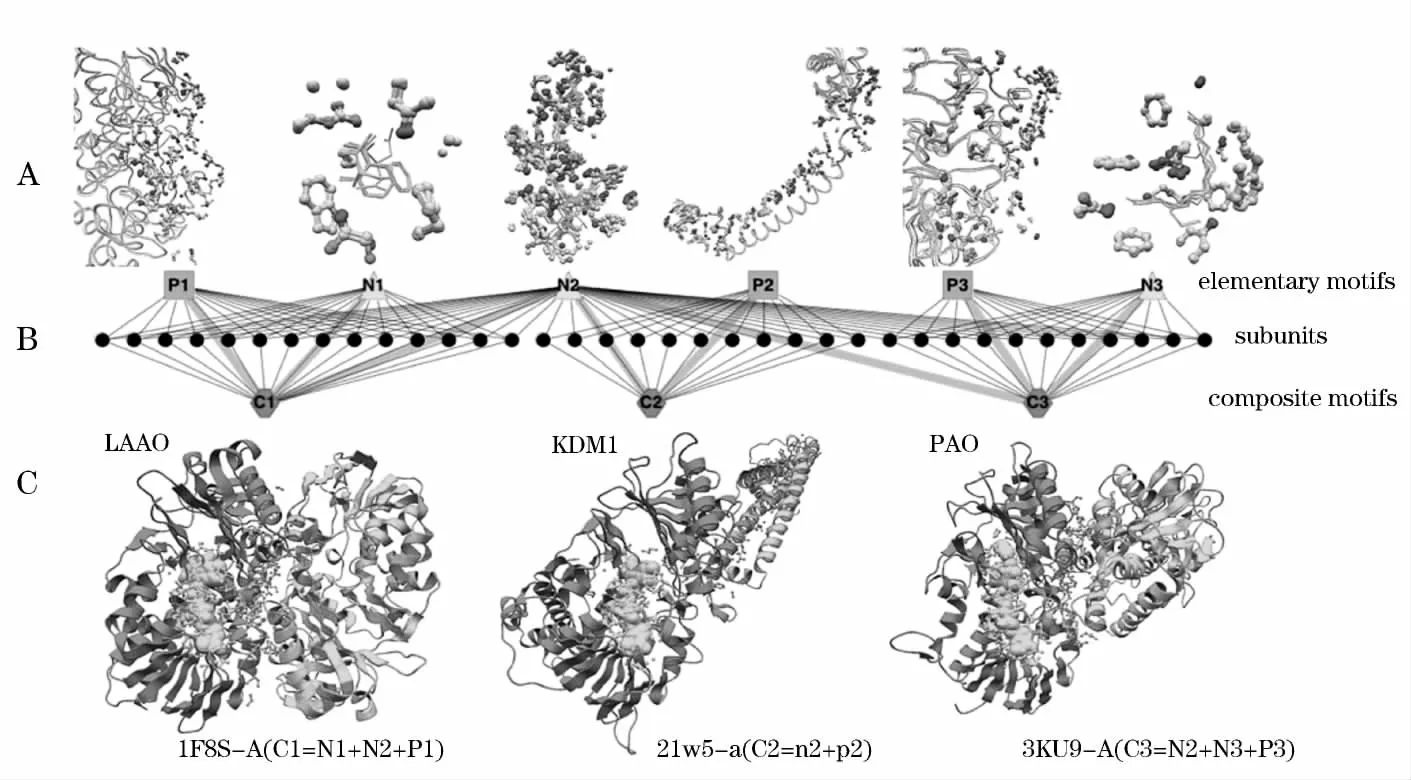
图6 基序通过完全连锁聚类得到复合基序Fig.6 Motif complete linkage clustering composite motif
Hoffmann[23]开发了一种衡量结合口袋之间相似性的新方法。以原子云代表每一个口袋,通过比对三维空间中的原子来评估两个口袋之间的相似性,并用convolution kernel比较所得到的结果信息。这样即使相关蛋白不共享序列和整体结构相似性,口袋比对也是可行的。并用此方法来识别已知的结合口袋的配体结合的相关性,为今后在这一领域的工作提供了新的标杆。Hermann[24]预测Tm0396的酶功能活性发现潜在的物的高能量结构对接模式可能成为酶功能预测的有用工具。
3.4 其他预测方法
Liao[25]建立了一种不依赖于序列和结构相似性来预测蛋白质功能的新方法。选择酵母中已知的实验测定的1377个蛋白质。首先将它们由短到长重新排列成一个连贯的数据集。设定一个连贯序列集m(可随机取值),将氨基酸序列集转换为profile编码(每个氨基酸在1377个总数中出现的频率)数据集。然后采用最邻近聚类算法对序列集进行测试。选择步长为5,设定m值,得到的结果30%m作为测试集,剩余作为训练集。这个方法是很多与已知功能序列相似性很小的新蛋白质序列得到预测,同时也增加了从序列预测功能的普及性。Yang[26]从序列的数字特征预测蛋白质功能。首先从序列中提取疏水性、极性与电荷特性三个数字特征,并提出序列功能可能性。然后综合特征向量和功能可能性,应用k-最近邻居算法(KNN)进行蛋白质的功能预测。该方法综合考虑了局部和全局信息,预测结果比基于序列相似性的方法更有效。
4 总结
近几十年来,蛋白质功能预测的方法不断被充实完善。本文仅指列出了部分有代表性的常用的蛋白质功能预测方法,但其中支持各个方法的算法本文就不多做陈述。后基因组时代的快速发展给我们带来机遇的同时也带来了巨大的挑战,蛋白质序列与结构的悬殊差异使我们不得不加快透彻分析序列的脚步,发展从序列预测蛋白质结构与功能的普遍性与准确性并存的方法就变得刻不容缓。而目前所提出的基于序列预测的方法还远远不能满足科学发展的要求。
References)
[1] T.Hawkins,M.Chitale and D.Kihara.New paradigm in protein function prediction for large scaleomics analysis[J].Mol.Biosyst,2008,4:223 –231.
[2] A.Al- Shahib,R.Breitling,DR.Gilbert.Predicting protein function by machine learning on amino acid sequences–a critical evaluation[J].BMC Genomics,2007,78:1 – 10.
[3] P.Bork,T.Dandekar,Y.Diaz- Lazcoz,F.Eisenhaber,M.Huynen and YP.Yuan.Predicting Function:From Genes to Genomes and Back [J].J.Mol.Biol,1998,283:707 -725.
[4] SF Altschul,TL.Madden,AA.Sch ffer,JH.Zhang,Z.Zhang,W.Miller and DJ.Lipman.Gapped BLAST and PSI-BLAST:a new generation of protein database search programs[J].Nucleic Acids Res,1997,25:3389 – 3402.
[5] B.Rost,J.Liu,R.Nair,KO.Wrzeszczynski and Y.Ofran.Automatic prediction of protein function [J].Cellular and Molecular Life Sciences,2003,60:2637 -2650.
[6] B.Rost.Enzyme function less conserved than anticipated[J].J Mol Biol,2002,318:595 – 608.
[7] B.Louie,R.Higdon,E.Kolker.A statistical model of protein sequence similarity and function similarity reveals overly-specific function prediction[J].PLoS One ,2009,4:e7546.
[8] T.Hawkins,S.Luban,D.Kihara.Enhanced automated function prediction using distantly related sequences and contextual association by PFP[J].Protein Sci.,2006,15:1550 – 1556.
[9] T.Hawkins,M.Chitale,S.Luban,D.Kihara.PFP:automated prediction of gene ontology functional annotations with confidence scores using protein sequence data[J].Proteins,2009,74:556 –582.
[10] I.Friedberg,M.Jambon,A.Godzik.New avenues in protein function prediction[J].Protein Sci,2006,15:1527 – 1529.
[11] G.Lopez,A.Rojas,M.Tress,A.Valencia.Assessment of predictions submitted for the CASP7 function prediction category[J].Proteins,2007,69:165 –174.
[12] M.Chitale,T.Hawkins,C.Park and D.Kihara.ESG:extended similarity group method for automated protein function prediction[J].BMC,2009,14:1739 -1745.
[13] GD.Bader,MP.Cary,C.Sander.Pathguide:a pathway resource list[J].Nucleic Acids Res,2006,34:D504 - 506.
[14] Chua HN,Sung WK,Wong L.Exploiting indirect neighbours and topological weight to predict protein function from protein-protein interactions[J].Bioinformatics,2006,22:1623.
[15] L.Hu,T.Huang,X.Shi,WC.Lu,YD.Cai,KC.Chou.Predicting Functions of Proteins in Mouse Based on Weighted Protein-Protein Interaction Network and Protein Hybrid Properties[J].PLOS ,2011,1:e14556.
[16] Chi and Hou:An iterative approach of protein function prediction[J].BMC Bioinformatics,2011,12:437.
[17] AW.Rives,T.Galitski.Modular organization of cellular networks[J].Proceedings of the National Academy of Sciences,2003,100:1128.
[18] J.Dutkowski,T.Ideker.Protein Networks as Logic Functions in Development and Cancer[J].PloS Computational Biology ,2011,9:e1002180.
[19] S.Goldsmith - Fischman,B Honig.Structural genomics:computational methods for structure analysis[J].Protein Sci,2003,12:1813-1821.
[20] GA Reeves,JM Thornton.Integrating biological data through the genome[J].Human Molecular Genetics,2006,7:R81 - R87.
[21] LC.James,AH.Keeble,Z.Khan,DA.Rhodes and J.Trowsdale.Structural basis for PRYSPRY -mediated tripartite motif(TRIM)protein function[J].PNAS,2007,104(15):6200 -6205.
[22] AR.Kinjo,H.Nakamura.Composite Structural Motifs of Binding Sites for Delineating Biological Functions of Proteins[J].PLoS ONE,2012,7(2):e31437.
[23] B.Hoffmann,M.Zaslavskiy,Jean - Philippe Vert and V.Stoven.A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D:application to ligand prediction.BMC Bioinformatics 2010,11:99.
[24] JC.Hermann,R.Marti-Arbona,AA.Fedorov,E.Fedorov,SC.Almo,BK.Shoichet and FM.Raushel.Structure-based activity prediction for an enzyme of unknown function[J].Nature,2007,448(7155):775 –779.
[25] B.Liao,Q.Liu,Q.Zeng,J.Luo,G.Yue.An Approach for Data Selection of Protein Function Prediction[J].MATCH Commun.Math.Comput.Chem,2011,65:459 -468.
[26] A.Yang,R.Li,W.Zhu,G.Yue.A Novel Method for Protein Function Prediction Based on Sequence Numerical Features[J].MATCH Commun.Math.Comput.Chem,2012,67:833 -843.
