木质素生物合成酶CCR基因的生物信息学分析
2013-11-14徐秉良白江平
陈 刚,徐秉良*,白江平
(1.甘肃农业大学草业学院、草业生态系统教育部重点实验室、中-美草地畜牧业可持续发展研究中心、甘肃省草业工程实验室,甘肃兰州730070;2.甘肃省干旱生境作物学重点实验室-甘肃省作物遗传改良与种质创新重点实验室,甘肃兰州730070)
木质素是一种具有芳香族特性的三维高分子化合物。作为地球上含量仅次于纤维素的天然有机物,木质素具有重要的生理功能,特别是在植物抗倒伏、抗病和抗逆境方面发挥着重要的作用[1]。木质素在植物体内的生物合成途径尚不完全清楚,但普遍认为大致包括莽草酸途径、苯丙氨酸途径及木质素特异合成途径3个主要阶段,目前对于木质素生物合成途径的研究越来越多的集中在木质素的特异合成途径上[2]。
肉桂酰辅酶 A还原酶(CCR)作为催化木质素特异途径的第一个关键酶,催化3种羟基肉桂酸的CoA酯还原生成相应的肉桂醛,可能对木质素合成途径的碳流具有潜在的调控作用,是调节碳素流向木质素潜在的控制关节点,对木质素单体的生物合成起着重要作用[3]。因此对CCR的研究将有助于对植物木质素生物合成途径的进一步研究。
到目前为止,已从拟南芥、大麦、小麦、番茄等多种植物中克隆得到了CCR基因的全长或部分编码序列[4],但对CCR基因缺乏系统的生物信息学分析和研究报道,特别是CCR基因编码氨基酸序列的保守区域、CCR蛋白导肽、信号肽、亚细胞定位、跨膜结构域、功能位点及三级结构的研究尚未见报道。为此,本研究拟采用生物信息学工具与分析方法,对NCBI数据库中分别来自裸子植物、单子叶植物及双子叶植物的35条CCR基因完整cDNA及其编码的氨基酸序列进行数据挖掘,旨在为对CCR基因的进一步研究和利用提供一定的理论依据。
1 材料与方法
1.1 材料
数据资料来源于NCBI数据库中已注册的,分别来自裸子植物、单子叶植物及双子叶植物共计35条CCR基因的核酸及其编码的氨基酸序列(表1)。
1.2 方法
利用NCBI中的ORF Finder和BioXM 2.6软件对CCR基因完整cDNA序列的GC含量进行分析;采用ClustalX和Mega4软件构建CCR基因的系统发生树;通过NCBI的Conserved Domains数据库,对CCR基因编码的氨基酸序列进行保守区分析;采用ExPASy、SMART、Post Prediction、TargetP 1.1 Server、SignalP 4.0 Server、TMHMM Server v.2.0、ProtScale及Cn3D对CCR基因编码的主要氨基酸的平均含量、理化性质、CCR蛋白结构域、亚细胞定位、导肽、信号肽、跨膜结构域、亲/疏水性以及CCR基因编码的氨基酸的活性位点、NADP结合位点及底物结合位点进行预测和分析;最后采用Swiss-Model对CCR基因编码蛋白质的三级结构进行同源建模,并用PyMOL对建模结果进行处理。
2 结果与分析
2.1 CCR基因完整cDNA序列的分析
2.1.1 CCR 基因 GC含量的分析
采用 NCBI中 ORF Finder和 BioXM 2.6对 35条CCR基因完整cDNA序列进行GC含量分析[5](表2),结果表明,单子叶植物CCR基因的GC含量,尤其是编码区GC含量远高于双子叶植物。单子叶植物中甘蔗CCR的GC含量最高,达69.89%;黑麦草CCR2的GC含量最低,为61.34%,平均为66.92%;而双子叶植物中GC含量最高为大叶相思,达 53.71%,苜蓿的最低,为 41.27%,平均48.84%。
2.1.2 CCR基因系统发育树的构建
采用ClustalX程序对35条CCR基因的完整cDNA序列进行多重比对(采用默认的IUB记分矩阵),采用Mega4程序对产生的多重比对结果构建系统发育树(采取最大简约法),并采用随机逐步比较的方式搜索最佳系统树,对生成的系统发育树进行Bootstrap校正,最终生成系统发育树[6](图1)。
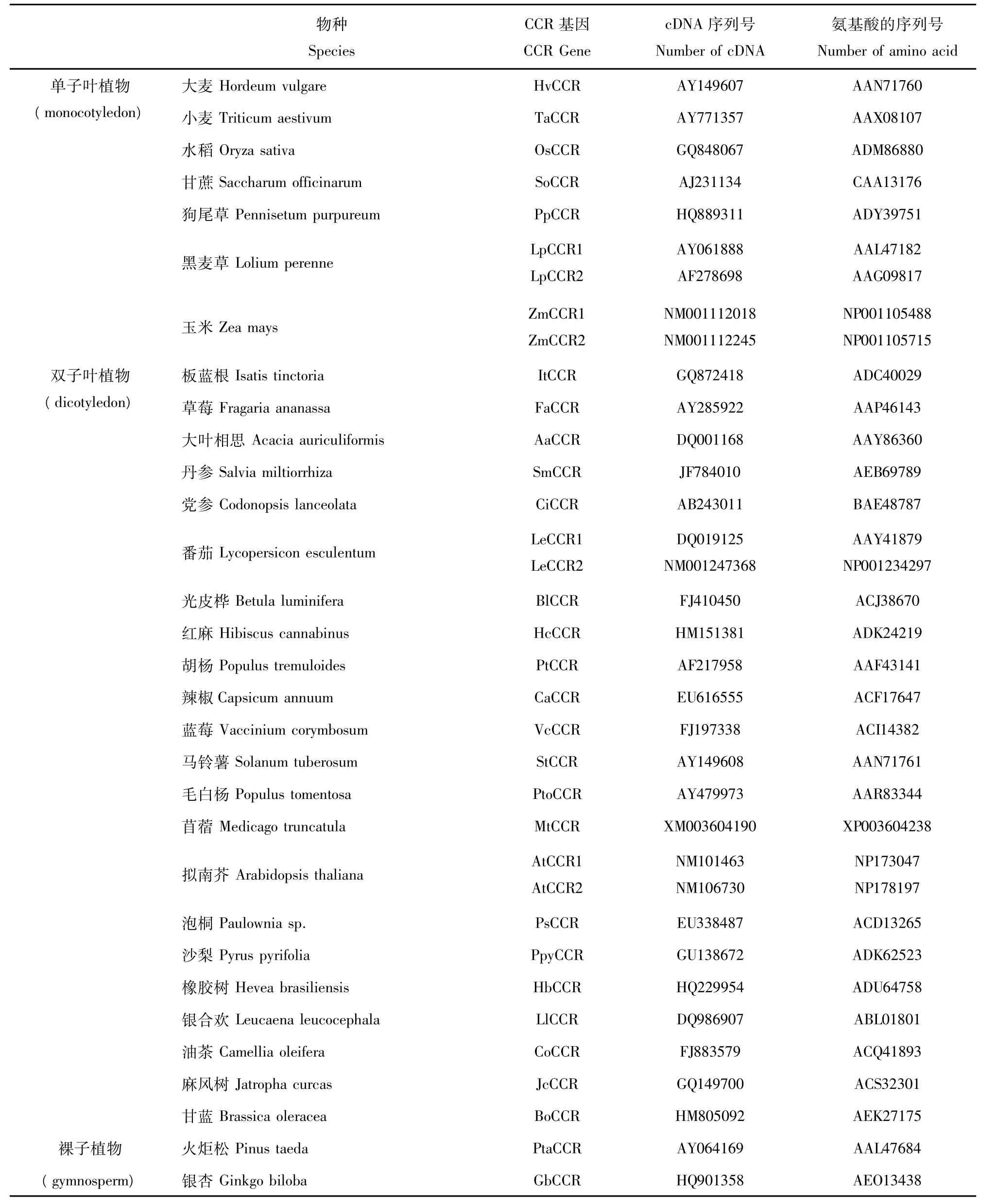
表1 不同植物CCR基因cDNA及其编码氨基酸的序列号Table1 The sequence number of cDNA and the corresponding amino acid of CCR in different plants物种
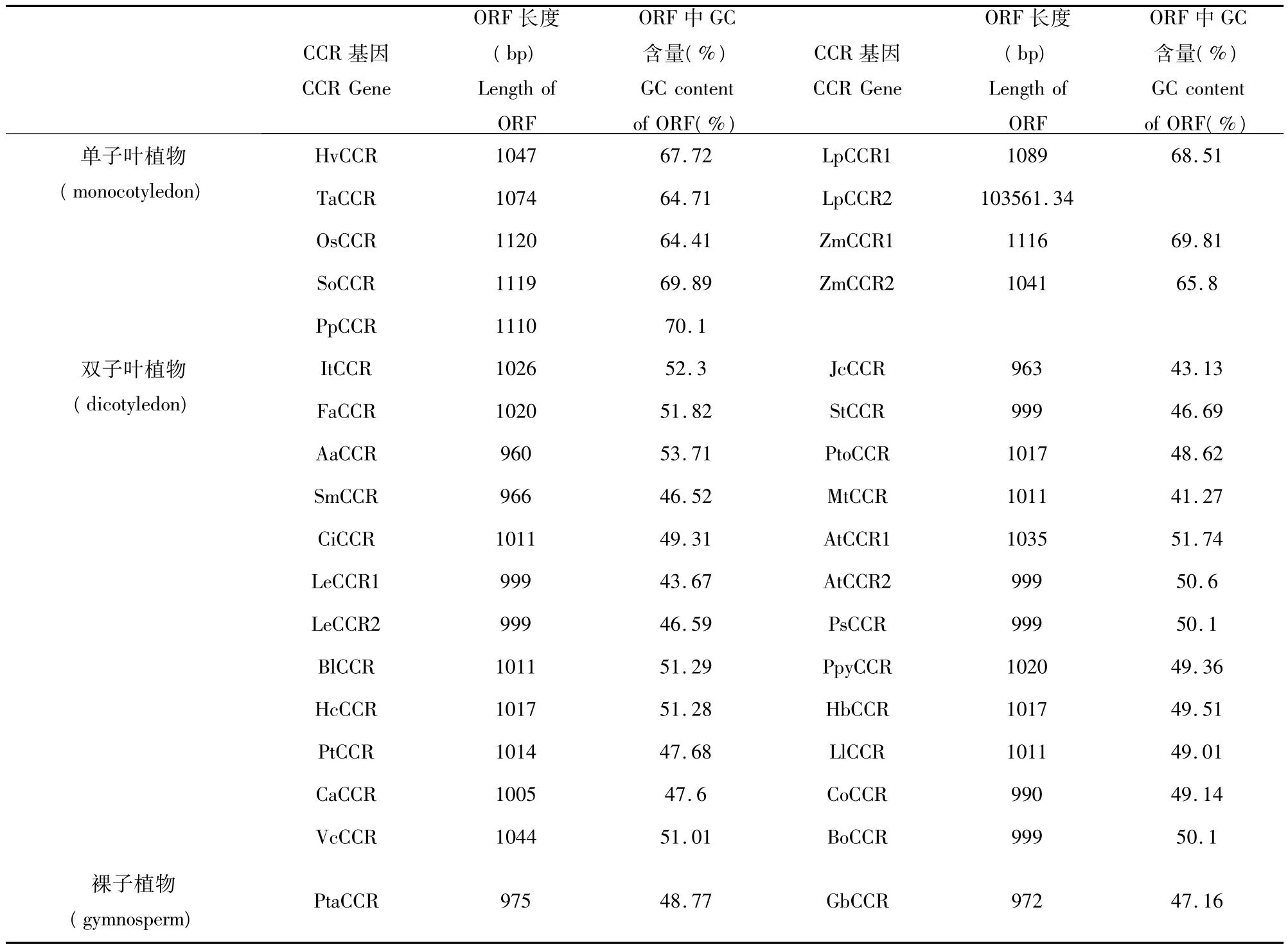
表2 CCR基因ORF长度及GC含量Table 2 The ORF length and GC content of CCR geneCCR基因
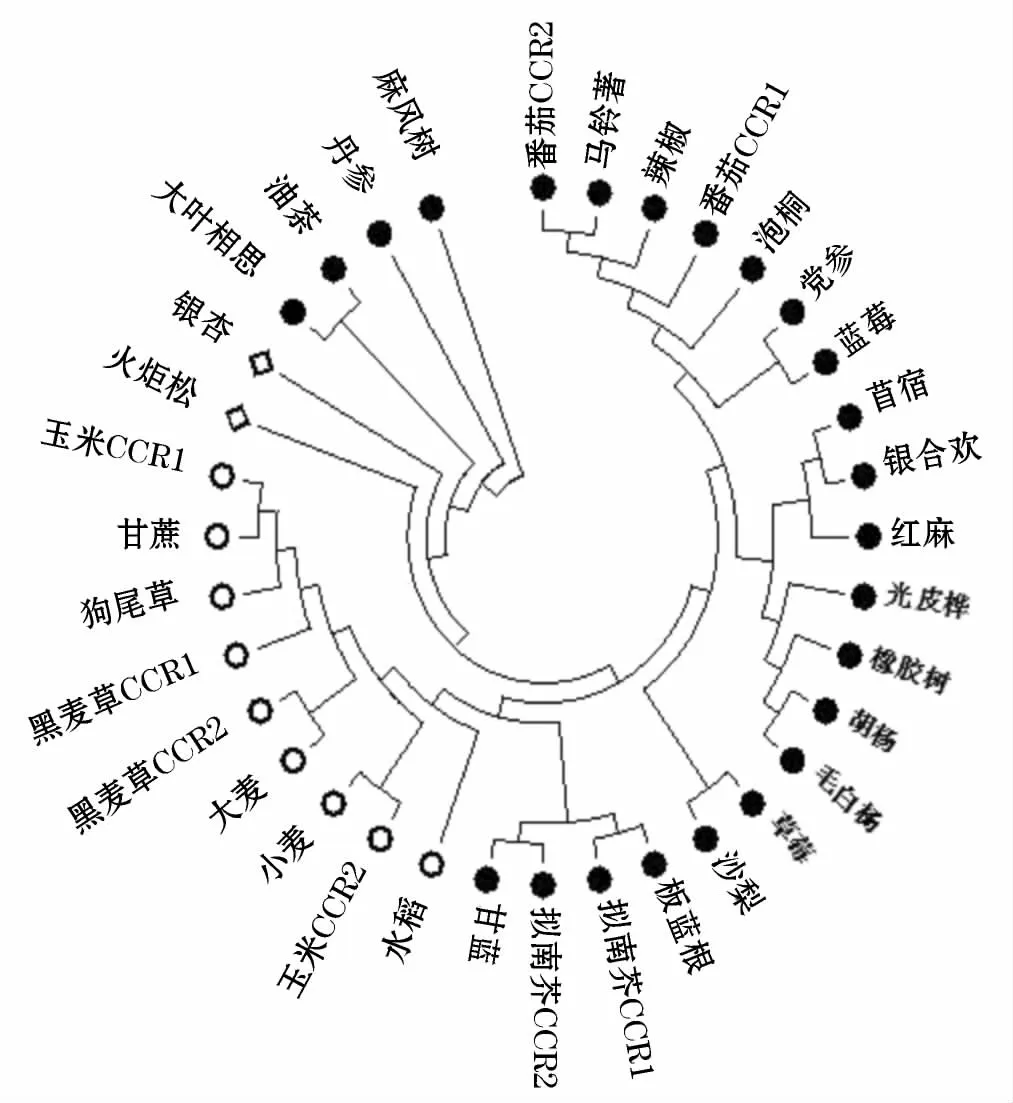
图1 CCR基因系统发育树Fig.1 Phylogenetic tree of CCR gene
由图1可以看出,植物CCR基因在进化树中大致可分为二大类、三小类,这与植物在进化的过程中分化为被子植物和裸子植物,被子植物又进一步分化成单子叶植物和双子叶植物的进化方式相一致,但双子叶植物中的拟南芥、板蓝根和甘蔗的CCR基因与单子叶植物的CCR基因聚在一类,麻风树、丹参、油茶及大叶相思的CCR基因与裸子植物银杏,火炬松的CCR基因聚在一类,其余双子叶植物的CCR基因全部聚在一类中。这表明在植物分化之前,CCR基因已经存在于植物中,而且在植物的进化时间上超前于植物的分化时间。在分类地位上,虽然拟南芥、板蓝根、甘蔗与单子叶植物差异较大,但其CCR基因与单子叶植物的CCR基因聚在同一类中,麻风树、丹参、油茶及大叶相思的CCR基因和裸子植物的CCR基因关系较近,这种基因的聚类和植物分类存在冲突的现象在植物中已被广泛发现[7]。通过构建 CCR基因系统发育树,CCR基因的聚类与植物的分类大体一致,表明CCR基因的进化和植物的进化基本是一致的,CCR基因和植物的进化过程密切相关。
2.2 CCR基因编码氨基酸保守区域及理化性质
2.2.1 CCR基因编码氨基酸序列保守区的分析
采用ClustalX(v1.83)软件对CCR基因编码氨基酸序列的保守区域进行分析[6],结果表明,从蛋白质的N端到C端,依次发现了以下9个氨基酸保守区(图略):①VCVTGAGGFIASWLVKLLL;②GYTVKGTVRNP;③GVFHTASP;④VTDDPEQMVEPAV;⑤VRRVVFTSSIGAV;⑥TKNWYCYGKAVAE;⑦GVDLVVVNPVLVIGPLLQ;⑧ASGRYLCAE;⑨TVKSLQEKGHL。在 NCBI的 Conserved Domains数据库中,对上述9个保守区域进行分析[6],结果表明9个保守区域共同构成了NADB_Rossmann Superfamily氨基酸保守区,功能注释为 Rossmannfold NAD(P)(+)-binding proteins,在反应中起催化还原的作用。
2.2.2 CCR基因编码主要氨基酸平均含量的分析
利用ExPASy ProtParam对35条CCR基因编码的含量较高的氨基酸进行统计[8],发现不论单子叶植物、双子叶植物、还是裸子植物,CCR基因编码的含量最高的氨基酸均为Ala、Val及Leu(麻风树中包括Ala和Val),但单子叶、双子叶植物中以Val的平均含量最高,裸子植物中中以Leu的平均含量最高。植物CCR基因编码的含量较高的氨基酸在单子叶植物中为:Val(11.83%) >Ala(11.79%) >Leu(8.58%) > Asp(6.56%) > Gly(6.52%);双子叶植物中为:Val(10.20%) > Leu(9.40%) > Ala(8.45%) > Lys(7.47%) > Glu(6.55%);裸子植物中为:Leu(10.25%) > Val(10.05%) > Ala(8.65%)> Lys(7.90%) > Glu(6.20%)=Gly(6.20%),其中 Val、Ala、Leu,Gly 均为非极性氨基酸,Glu、Asp 均为酸性氨基酸,Lys为碱性氨基酸。
对木质素生物合成途径中另外两种基因4CL和C3H所编码的主要氨基酸进行统计分析,结果表明,不论在单子叶植物、双子叶植物还是裸子植物中,4CL和C3H所编码的最主要的氨基酸均为Val、Ala 与 Gly[9-10],均属非极性氨基酸,与 CCR 编码的主要氨基酸的种类一致,但4CL、C3H及CCR所编码的含量最高的氨基酸的种类并不相同,CCR编码的含量最高的氨基酸为Val,4CL编码的含量最高的氨基酸为Ala,而C3H编码的含量最高的氨基酸为Leu。
2.2.3 CCR基因编码氨基酸理化性质的分析
采用ExPASy ProtParam对35条CCR基因编码氨基酸的理化性质进行分析[8],结果表明,不同植物CCR基因编码的氨基酸残基数、分子质量、酸性/碱性氨基酸比例、蛋白质不稳定性指数基本一致。在蛋白质的稳定性中,除水稻的CCR蛋白为不稳定蛋白外,其他植物的CCR蛋白均为稳定性蛋白。
2.3 CCR蛋白结构和功能的预测及分析
2.3.1 CCR蛋白导肽、信号肽的预测和分析
导肽是一段新合成的肽链携带的通过细胞膜进入细胞器的所必须的识别序列。采用TargetP 1.1 Server对35条CCR基因编码的氨基酸序列进行预测和分析[11],结果表明其序列含叶绿体转运肽及线粒体目标肽的分值均较低,无氨基酸残基裂解位点,可靠性Ⅳ级,说明CCR蛋白不存在上述2种导肽。
信号肽位于蛋白质的N端,指导分泌性蛋白到内质网膜上合成,在蛋白质合成结束之前被切除,一般有16~26个氨基酸残基,其中包括疏水核心区、信号肽的C端和N端。采用SignalP 4.0 Server对CCR蛋白的信号肽进行分析[12],结果表明,植物CCR蛋白无信号肽。
2.3.2 CCR蛋白跨膜结构域、亚细胞定位的预测和分析
跨膜结构域通常由20个左右的疏水性氨基酸残基组成,主要形成α螺旋。采用TMHMM Server v.2.0软件对35条CCR基因编码氨基酸的跨膜结构域进行预测和分析[13]。结果表明,CCR蛋白不存在跨膜蛋白,大部分蛋白质位于膜内,但有一部分氨基酸残基的肽段嵌入膜内。
对35条CCR基因编码的氨基酸采用Post Prediction进行亚细胞定位[14],结果表明,65.7% 的CCR蛋白定位于质膜上、22.9%的定位于细胞质中、8.6%的定位于内质网上,另有2.9%的定位于细胞核上。由此推断,在不同的植物体中,虽然CCR蛋白的定位有所不同,但绝大多数定位于质膜上,少数定位于细胞质中。
2.3.3 CCR蛋白亲/疏水性的预测及分析
采用ExPASy ProtScale对35条CCR基因编码的氨基酸序列分析[11],结果表明,其氨基酸序列中亲水性氨基酸、疏水性氨基酸均匀的分布于整个肽链中,亲水性氨基酸多于疏水性氨基酸,因此可认为CCR蛋白属于亲水性蛋白。
2.3.4 CCR基因编码蛋白结构域的预测及分析
在NCBI的Conserved Domains数据库中,对35条CCR基因进行分析,结果表明,与提交的序列最匹配的保守结构域模型为FR_SDR_e,其在相同的蛋白质序列中生成的重叠为NADB_Rossmann Superfamily(功能注释为 Rossmann-fold NAD(P)(+)-binding proteins)。
同时,采用SMART对CCR蛋白氨基酸序列的功能结构域进行分析[15],结果表明,在蛋白的N端存在一个脱氢酶/差向异构酶/NAD结合蛋白的结构域,即与3Beta_HSD/Epimerase/NAD_binding_4等保守域具有很高的同源性(图2)。
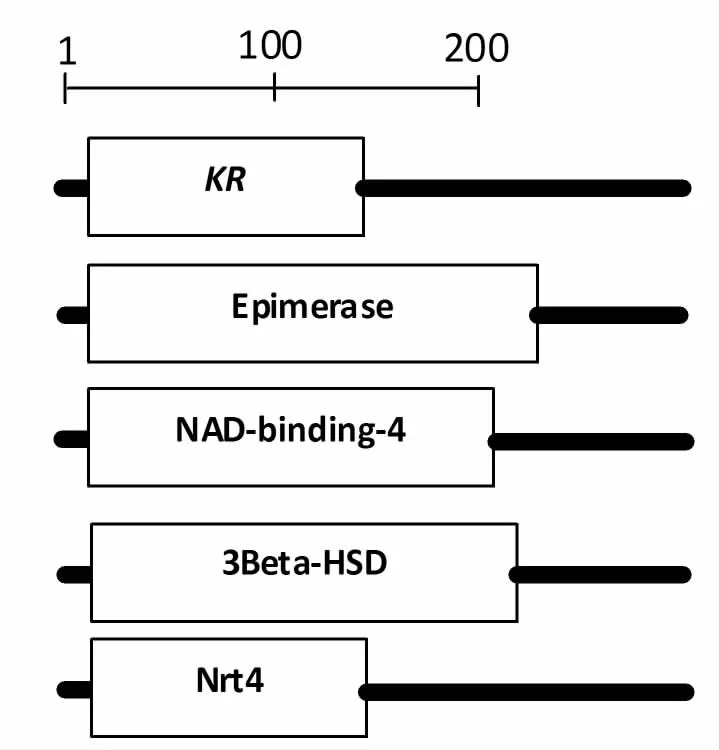
图2 CCR蛋白结构域的预测Fig.2 Predicted domain sites of CCR
2.3.5 CCR蛋白活性位点、NADP结合位点及底物结合位点的预测和分析
酶的特殊催化能力只局限在大分子的一定区域,只有少数特殊的氨基酸残基参与底物结合及催化作用,这些特异的氨基酸残基比较集中的区域,即与酶活力直接相关的区域称为酶的活性部位。酶的活性部位通常又分为结合部位和催化部位[5]。为了进一步了解CCR蛋白活性位点、NADP结合位点及底物结合位点在CCR中的分布,利用Cn3D对FR_SDR_e模型进行分析[16](图3),结果表明拟南芥CCR1的活性位点分别位于第98位(A)、第122位(S)、第156位(Y)和第160位(K);NADP结合位点分别位于第12位(G)、第14位(G)、第15位(G)、第16位(F)、第17 位(I)、第36 位(V)、第 37位(R)、第62位(A)、第63位(D)、第 64 位(L)、第83位(T)、第84位(A)、第85位(S)、第86位(P)、第87位(M)、第120位(T)、第121位(S)、第156位(Y)、第 160位(K)、第 183位(P)、第 184位(V)、第185位(L)、第186位(V)和第198位(S);底物结合位点分别位于第87位(M)、第89位(D)、第122位(S)、第123位(I)、第124位(G)、第127位(Y)、第 156位(Y)、第 183位(P)、第 184位(V)、第185 位(L)、第198 位(S)、第201 位(H)、第215位(N)、第219位(V)和第285位(F)。
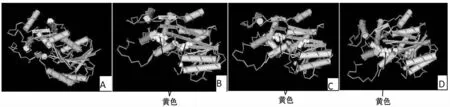
图3 CCR蛋白活性位点、NADP结合位点及底物结合位点的预测A:FR_SDR_e模型;B:黄色表示活性位点;C:黄色表示NADP结合位点;D:黄色表示底物结合位点。Fig.3 The actice sites,NADP-building sites and substrate-building sites of CCR protein A:The model of FR_SDR_e;B:Yeoolow show active sites.C:Yellow shows NADP binding sites.D:Yellow shows substrate binding sites.
2.3.6 CCR蛋白三级结构的预测和分析
蛋白质要实现其催化等活性首先要正确的完成折叠,因此对蛋白多肽构成以及一级结构的分析远远不能满足对蛋白酶功能的了解。蛋白质三级结构的分析,对理解蛋白质结构和功能之间的关系起了至关重要的作用。目前的X-ray和NMR等实验技术预测蛋白质的结构代价相当高,随着生物信息学的发展,用生物软件预测蛋白质的结构变成现实[17]。采用Swiss-Model对植物CCR基因编码蛋白质的三级结构进行同源建模,并用PyMOL对建模结果进行处理[18]。结果表明CCR蛋白的三维结构以α-螺旋和无规卷曲为主要的结构元件,延伸链分布于整个肽链之中(图4)。
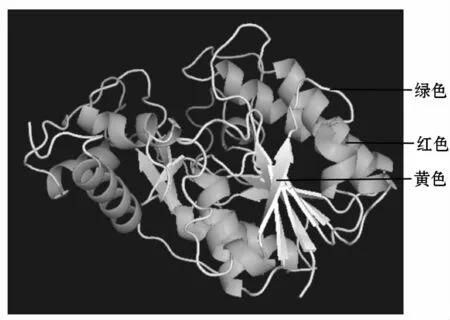
图4 CCR蛋白三维结构模型的预测红色:α-螺旋;黄色:β-折叠延伸链;绿色:无视卷曲Fig.4 Three-dimensional structure prediction of CCR protein Red:Alpha helix;Yelllow:Beta sheet extended strand;Green:Random coil
采用Swiss-Pdb Viewer分析拟南芥CCR1的同源建模结果,结果表明,预测的蛋白质残基的二面角(ψ和φ)位于黄色核心区域(图5),表明其空间结构稳定,所以用同源建模的方法对植物CCR基因编码的氨基酸序列进行上述建模的结果非常可靠。
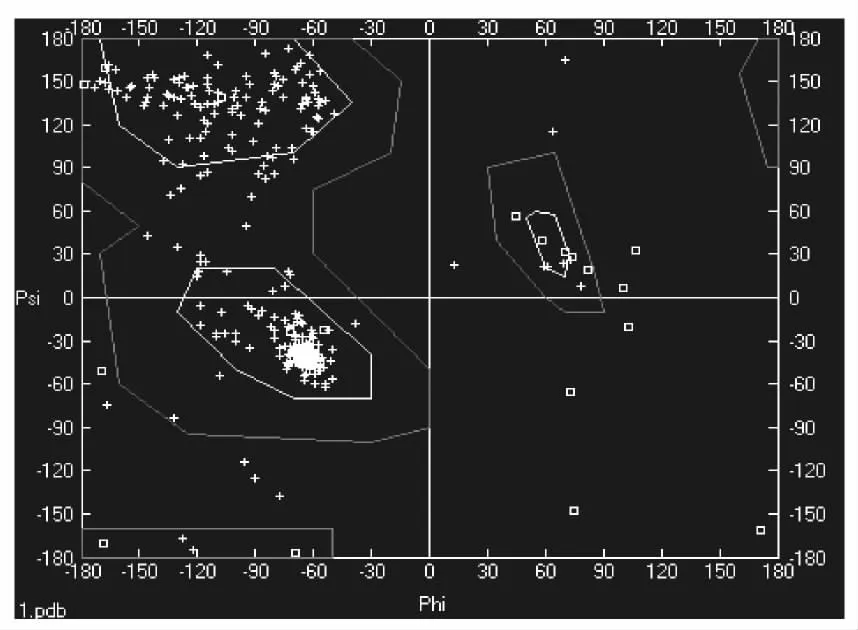
图5 拟南芥CCR1蛋白模型的拉氏构象图Fig.5 Ramachandran plot prediction of CCR1 protein in Arabidopsis thaliana
3 结论与讨论
本研究应用生物信息学手段,对NCBI数据库中分别来自裸子植物、单子叶植物及双子叶植物的35条CCR基因完整cDNA及其编码氨基酸序列的组成成分、理化性质、保守序列、导肽、信号肽、跨膜结构域、亲/疏水性,结构域及CCR蛋白的三级结构进行了预测和分析,构建了CCR基因的系统进化树和CCR蛋白三级结构的模型。
分析结果表明,单子叶植物CCR基因GC含量,尤其是编码区的GC含量较双子叶植物的普遍偏高,这种现象不仅在CCR基因中如此,在对其他基因的研究中亦有发现[9-10],因此推测高的 GC含量可能是单子叶植物基因区别于双子叶植物基因的一个典型特征。CCR基因GC含量在两大类植物中的明显差异可能与植物的进化过程和生存环境的差异有一定联系[9];CCR基因的进化与植物的进化基本一致,但少数CCR基因的聚类和植物分类存在冲突,有研究表明基因的倍增和重组、水平的基因转移等都是这种差异存在的原因[10];CCR基因编码的氨基酸从N端到C端依次发现了9个保守区域,在反应中共同起催化还原的作用,但目前大多数的文献中对KNWYCYGK这一保守区域报道较多,且认为这一区域在超二级结构上可形成βαβ结构,并推测它可能是CCR的催化位点,也可能是其辅因子NADPH的结合区域,尤其是其上的两个赖氨酸残基(K)可能直接与底物结合,但本文通过对已报道的35条CCR蛋白二级结构以及上述功能位点的分析发现KNWYCYGK在超二级结构上并不能形成βαβ结构,仅能形成一段α-螺旋和部分无规卷曲,其中仅有Y(N-端)、K(C-端)与CCR蛋白的催化位点、NADPH结合位点及底物结合位点K(C-端)有关。
CCR基因编码氨基酸的理化性质基本一致,但不同植物中CCR基因编码的主要氨基酸的种类和含量存在着差异;CCR基因与木质素合成过程中C3H、4CL基因与所编码的主要氨基酸种类相同,均为Val、Ala及Gly,因此推测这3种氨基酸可能与木质素合成过程中相关的酶有重要联系;不同植物CCR基因编码的氨基酸残基数、分子质量、酸性/碱性氨基酸比例、蛋白质不稳定性指数基本一致;CCR蛋白无导肽、信号肽及跨膜结构域,属亲水性蛋白;主要定位于质膜上,少数定位于细胞质中,且通过对CCR蛋白跨膜结构的预测和分析结果可知,定位于质膜上的蛋白质主要以外在蛋白或脂锚定蛋白的形式存在,少数以整合蛋白的形式部分嵌入质膜中,另外,CCR蛋白质在核糖体上合成后,可能并不进行蛋白转运,而是直接与质膜结合,或保留在细胞质基质中起催化还原的作用;CCR蛋白的N端存在一个脱氢酶/差向异构酶/NAD结合蛋白结构域,是其进行催化还原反应的主要部位;CCR蛋白三级结构模型的空间结构稳定,建模结果可靠。分析结果对于深入研究CCR蛋白在木质素合成中的作用具有一定的指导意义。
References)
[1] Hano C,Addi M,Bensaddek L.Differential accumulation of monolignol-derived compounds in elicited flax(Linum usitatissimum)cell suspension cultures[J].Planta,2006,223(5):975 -989.
[2] 耿飒,徐存拴,李玉昌.木质素的生物合成及其调控研究进展[J].西北植物学报,2003,23(1):171 -181.
[3] Lacombe E,Hawkins S.Cinnamoyl CoA reductase,the first committed enzyme of the lignin branch biosynthetic pathway:cloning,expression and phylogenetic relationships[J].Plant Journal,1997,11(3):429 -441.
[4] 李金花,张绮纹,牛正田,卢孟柱,Carl J Douglas.木质素生物合成及其基因调控的研究进展[J].世界林业研究,2007,20(1):29-37.
[5] 薛庆中主编,DNA和蛋白质序列数据分析工具[M].第二版.北京:科学出版社,2009,72 -100.
[6] Kumar S,Tamura K,Nei M.Integrated software for molecular evolutionary genetics analysis and sequence alignment[J].Briefings in Bioinformatics,2004,5:150 -163.
[7] Doyle J J.Trees within trees:genes and species,molecules and morphology[J].Syst Biol,1997,46:537 -553.
[8] Kyce J,and Doolittle RF.A simple method for displaying the hydropathic character of a protein[J].Mol Biol,1982,157(6):105-132.
[9] 黄胜雄,胡尚连,孙 霞,曹 颖,卢学琴,蒋 瑶.木质素生物合成酶4CL基因的遗传进化分析[J].西北农林科技大学学报(自然科学版),2008,36(10):199 -206.
[10] 薛永常,聂会忠,刘长斌.木质素合成酶C3H基因的生物信息学分析[J].生物信息学,2009,7(1):13-17.
[11] Emanuelsson O,Nielsen H,and Brunak S.Predicting subcellular localization of proteins based on their N-terminsl smino acid sequence[J].Mol Biol,2000,300(4):1005 -1016.
[12] Bendtsen J D,Nielsen H and Von Heijne G.Improved prediction of signal peptides:SingalP 4.0 [J].Mol Biol.,2004,340(4):783-795.
[13] Iked A M,Arai M,and Lao D M.Transmembrane topology prediction methods:a reassessment and improvement by a consensus method using a dataset of experimentally characterized transmembrane topologies[J].In Silico Biol,2002,2(1):19 -33.
[14] 刘旭光,张 杰编著,分子生物学软件应用[M],第一版.北京:北京大学医学出版社,2007:178.
[15] Page R D M,Charleston M A.From gene to organismal phylogeny:reconciled trees and the gene tree/species tree problem[J].Mol Phylogenet Evol,1997,7:231 -240.
[16] 付海辉,辛培尧,许玉兰,刘 岩,韦援教,董 娇,曹有龙,周,军.几种经济植物UFGT基因的生物信息学分析[J].基因组学与应用生物学,2010,30(1):92 -102.
[17] Arnold K,Bordoli L,Kopp J,Schwede T.The SWISS - MODEL workspace:A web-based environment for protein structure homology modeling[J].Bioinformatics,2006,22(2):195 -201.
[18] Laskowski R A,Macarthur M W,Moss D S,Thornton J M.PROCHECK:A program to check the stereo chemical quality of protein structures[J].Appl.Cryst.,1993,26(2):283 -291.
