气相色谱-负化学电离质谱法测定沉积物和鱼肉中毒杀芬的8个同类物及其总含量
2013-10-22劳文剑
劳文剑
(南加州海岸水环境研究所,美国加利福尼亚州科斯塔梅萨市 92626)
毒杀芬是一种广谱的有机氯杀虫剂,主要由氯代莰烷和氯代莰烯同类物(或单体)组成。毒杀芬具有亲脂性,可以被生物富集,能够在环境中持久的存在。它的半挥发性有利于长距离的大气传输,因此目前已在土壤、水体、底泥、鱼和海洋哺乳动物体内检测到毒杀芬[1-3]。通常采用气相色谱法(GC)分析毒杀芬[4]。理论上毒杀芬可以有32768个同类物,但目前通过二维气相色谱只发现了约1000个同类物[5,6]。GC不能全部分离数量巨大的毒杀芬同类物,而且,有些同类物还和其他有机氯杀虫剂(如多氯联苯(PCB)和氯丹等)从色谱柱上共流出,这就使得分析毒杀芬成为一项富有挑战性的工作。测定毒杀芬的含量通常有两种方法:一是以工业毒杀芬(technical toxaphene)为标准样品估计毒杀芬的总含量(方法Ⅰ);二是用选定的纯同类物为标准样品测定少数毒杀芬同类物的含量(方法Ⅱ),进而估计毒杀芬的总含量,这一方法与测定其他单个化合物的方法并无太大差别。方法Ⅰ一方面受来源于不同厂家甚至同一厂家的工业毒杀芬组成和含量变化的影响很大;另一方面,工业毒杀芬一旦进入自然环境中会逐渐风化(weathering),导致组成和含量与作为标准样品的工业毒杀芬不同,更增加了毒杀芬总残留量的定量误差[7]。
为了更加准确地定量分析毒杀芬,在分析方法的3个层面(即在样品制备阶段进行干扰物的预分离、色谱分离技术和检测方法)上,研究人员进行了不断的改进。例如,在干扰物的预分离方面,Krock等[8]使用8.0 g活化硅胶去除PCB的干扰,Maruya等[9]则使用18.0 g含水量1%的弗罗里硅土去除鱼肉萃取液中PCB和脂类的干扰。在色谱分离技术方面,从非极性柱(如 DB-5)到中等极性柱(DB-1701)都有使用[4],特别是 Smalling 和 Maruya[10]发现弱极性柱DB-XLB柱对毒杀芬同类物有较好的分离能力。中心切割多维色谱和全二维色谱技术对毒杀芬同类物有着更加强大的分离能力[11,12],张兵等[13]使用 DB-XLB为一维柱、BPX-50为二维柱的组合开发了对毒杀芬同类物分析的全二维色谱方法。在检测方法方面,电子捕获检测器(ECD)、电子轰击离子源-质谱(EI-MS)、负化学电离源-质谱(NCI-MS)、串联质谱(EI-MS/MS,NCI-MS/MS)、飞行时间质谱(TOF-MS)、高分辨质谱(HRMS)等都有应用[5,9,14-21]。其中 NCI-MS 检测以其对毒杀芬同类物的高选择性、高灵敏性和相对低的仪器价格,有着普遍应用的可行性。然而,NCI-MS也有其缺点,即对毒杀芬同类物的响应因子差别很大,特别是会受到PCB氧反应产物的干扰[4]。所谓的PCB氧反应是指在NCI-MS的离子源中如存在痕量的氧气,会与PCB反应生成[M-Cl+O]-和[M-Cl2+O]-的碎片离子,这些碎片离子的质荷比(m/z)与对应的毒杀芬同类物的定量离子相同,从而对毒杀芬的测定造成干扰。有关PCB氧反应干扰的问题详见笔者最近发表的文章[22]。
在中国,毒杀芬的分析检测在多个行业受到重视。例如,早在1995年,国家进出口商品检验局就发布了出口水产品中毒杀芬残留量的检验方法[23]。该方法使用弗罗里硅土净化样品,GC-ECD检测,采用4个同类物峰面积的和来估算毒杀芬的含量。2005年,国家烟草专卖局发布了毒杀芬残留的GCECD 检测方法[24]。2006 年,王明泰等[25]报道了使用GC/EI-MS对纺织品中的毒杀芬残留进行检测的方法。2009年,谢原利等[26]开发了加速溶剂萃取结合GC/NCI-MS测定土壤中毒杀芬总量的方法。2010年,张兵等[27]建立了使用同位素稀释-气相色谱-串联质谱(ID-GC-MS/MS)分析土壤中3个毒杀芬同类物(P26、P50和P62)的方法。2012年,该小组[13]报道了分析土壤中23个毒杀芬同类物的全二维气相色谱方法(GC×GC-ECD)。同年,田绍琼等[28]报道了GC/EI-MS/MS法测定人参和黄芪中7个毒杀芬同类物的方法。此外,还有包含毒杀芬检测的综述发表[29,30]。
美国环境保护署(USEPA)已有的Method 8081使用GC-ECD分析毒杀芬[31]。由于该方法存在检测选择性差,易受其他化合物干扰的缺点,USEPA于2012年发表了新的方法(Method 8276)[32]。新方法基于GC/NCI-MS,可用于定量检测水体、沉积物(底泥)和生物(鱼类)样品中总工业毒杀芬和8个通常有高残留的毒杀芬同类物(Hx-Sed、Hp-Sed、P26、P41、P40、P44、P50 和 P62)的含量。笔者参与了由USEPA组织的该方法多实验室联合确证分析工作的全部3个阶段。由于Method 8276是新的方法,目前还缺乏与之适应的具有可操作性的实用方法。本研究以Method 8276为参考,结合样品制备技术,建立了实用的毒杀芬的GC/NCI-MS分析方法,并应用于检测环境样品(滨海河口地区的沉积物)和鱼肉样品中毒杀芬总量(总毒杀芬)和8个毒杀芬同类物的含量。
1 实验部分
1.1 仪器、试剂与材料
Agilent 7890型气相色谱仪,配备5975C质谱检测器(美国Agilent公司)。Dionex ASE 300型加速溶剂萃取仪(美国Dionex公司)。TurboVap 500型浓缩仪(美国Zymark公司)。
8个毒杀芬同类物标准样品购自美国RT Corp公司。工业毒杀芬标准样品购自美国Restek公司。PCB混合标准液(包括42个单体)和单标准溶液(PCB204和 PCB209),以及 4,4'-二溴八氟联苯(DBOFB)购自美国Accustandard公司。正己烷(农残级)、二氯甲烷(HR-GC级)、硅胶(60~200目)购自美国J.T.Baker公司。氧化铝(60~325目)购自美国Fisher Scientific公司。硅胶和氧化铝分别于160℃和250℃下活化12 h,然后加入3%(质量分数)的去离子水去活,保存在正己烷中备用。无水硫酸钠(美国Mallinckrodt公司)于500℃下烘焙4 h备用。凝胶渗透色谱(GPC)填料SX-3 Bio-Beads(美国Bio-Rad公司)。
沉积物采自美国加州蒂华纳河口(Tijuana River estuary)。三文鱼样品购自美国加州某超市。所有样品均于-20℃下保存。
1.2 样品制备
冷冻干燥后的沉积物(1~5 g干重)和三文鱼样品(2.5~5 g干重)分别装入加速溶剂萃取仪的34 mL不锈钢萃取池中,加入回收率指示物DBOFB和PCB209(每个25 ng,以使在定容后的样品萃取液中的目标质量浓度为50 μg/L)后,以二氯甲烷为溶剂,进行加速溶剂萃取(ASE)。萃取条件:100℃和10.3 MPa下静态萃取5 min,进行4次提取循环,然后用60%的萃取池体积的二氯甲烷冲洗萃取池,最后用高纯氮气吹扫萃取池100 s。所收集的提取液在旋转蒸发仪上浓缩至约3 mL,加入约5 mL正己烷,再浓缩,以置换提取液中的二氯甲烷,这样置换3次,得到以正己烷为溶剂的萃取液。对于沉积物的萃取液,加入活化铜粉去硫,室温下避光放置过夜。取3 μL×3三文鱼萃取液在微量天平上测定样品的脂肪含量,其余萃取液须经GPC净化。GPC玻璃柱(2.5 cm(内径)×50 cm(长))装填40 g SX-3 Bio-Beads。上样后,用二氯甲烷-正己烷(体积比为1∶1)混合液洗脱,收集第100至220 mL的洗脱液。该洗脱液用上述方法在旋转蒸发仪上浓缩、溶剂置换为正己烷,待进一步净化。
经脱硫的沉积物萃取液和经GPC净化的三文鱼萃取液进一步由硅胶和氧化铝复合玻璃柱(10 mm(内径)×30 cm(长))净化。该柱由下到上依次装填约1 cm的无水硫酸钠、12 cm的硅胶(约4.7 g)、6 cm的氧化铝(约5.7 g)和约1 cm的无水硫酸钠。上样后,先用15 mL正己烷洗脱,并全部收集之;然后使用60 mL二氯甲烷-正己烷(3∶7,体积比)混合液洗脱并收集。收集的洗脱液用上述方法在旋转蒸发仪上浓缩、溶剂置换为正己烷后,定容至0.5 mL,加入内标 PCB204(50 μg/L),待 GC/NCI-MS 测定[33,34]。
1.3 分析条件
色谱柱为DB-XLB(30 m×0.25 mm×0.25 μm);载气为氦气(纯度≥99.999%),流量为 1 mL/min;进样口温度280℃,不分流进样,进样量1 μL;升温程序:90℃保持1 min,以5℃/min速率升至150℃,以3℃/min速率升至260℃,再以20℃/min速率升至320℃,保持5 min。样品的分析时间是57 min。
质谱检测器用负化学电离源。反应气为甲烷(纯度≥99.99%),40%流量。电离源和四极杆温度150℃;传输线温度280℃;选择离子检测(SIM)模式;溶剂延迟8 min。
2 结果与讨论
2.1 分析条件
使用DB-XLB色谱柱可以很好地分离8个毒杀芬同类物(见图1)。表1列出了以PCB204为内标的8个毒杀芬同类物的相对保留时间(RRT),即目标化合物的保留时间与内标PCB204的保留时间的比值。进样这8个毒杀芬同类物和PCB的混合标准液发现,有几个毒杀芬同类物会与PCB单体共流出,例如,P26与PCB81,P40与PCB158,P41部分地与PCB138,P44部分地与PCB187共流出。这些共流出的色谱峰若以ECD检测,则无法分辨。然而,在NCI-MS的SIM模式下,除了PCB187有可能会因氧反应而干扰P44的定量外,其他共流出的PCB单体不会对相应的毒杀芬同类物产生影响。此外,P44还会受另一个保留时间相近的毒杀芬同类物P42拖尾的影响,因此,在识别和积分P44色谱峰时要特别注意。另一方面,这也就使得色谱峰保留时间的稳定性在分析毒杀芬同类物时显得更加重要。在本研究的色谱条件下,连续多次进样的相对保留时间的相对标准偏差(RSD)小于0.02%,显示了色谱峰保留时间的高稳定性。

图1 8个毒杀芬同类物校正标准溶液(200 μg/L)在选择离子检测模式下的GC/NCI-MS色谱图Fig.1 GC/NCI-MS chromatogram of the eight toxaphene congeners in calibration standard(200 μg/L)under the selected ion monitoring mode

表1 以PCB204为内标的8个毒杀芬同类物的相对保留时间(RRT)和平均相对响应因子(RRF)及相对响应因子(RRF)的相对标准偏差(RSD)Table 1 Relative retention times(RRTs),average relative response factors(RRFs),and relative standard deviations(RSDs)of the relative response factors(RRFs)of the eight toxaphene congeners with PCB204 as internal standard
毒杀芬在较低的离子源温度下产生较高的离子丰度[32,35]。例如,在本研究中,150 ℃的离子源产生的离子丰度是106℃的(79±4.8)%。但是106℃的离子源温度易受到从色谱柱来的高温载气的影响而产生波动,因此离子源的温度还是选择在150℃。
毒杀芬总量和8个毒杀芬同类物的GC/NCIMS分析采用同一套质谱参数。在SIM模式下的定性、定量分析离子见表2。毒杀芬在GC/NCI-MS的选择离子扫描模式下的总离子流图见图2。
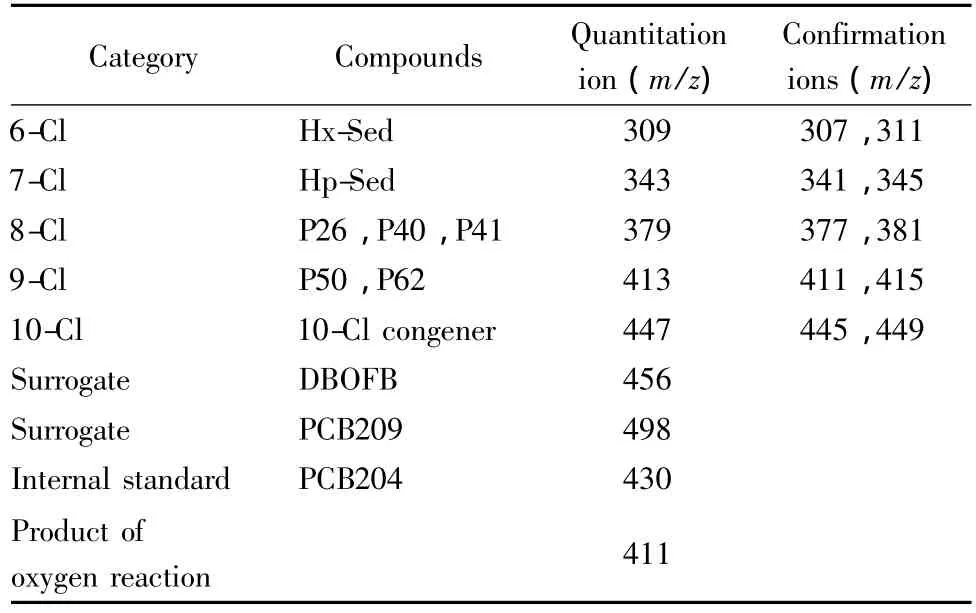
表2 GC/NCI-MS分析毒杀芬的定性和定量离子Table 2 Quantitation and confirmation ions for toxaphene analysis by GC/NCI-MS

图2 工业毒杀芬校正标准溶液(250 μg/L)在选择离子检测模式下的GC/NCI-MS色谱图Fig.2 GC/NCI-MS chromatogram of the technical toxaphene calibration standard(250 μg/L)under the selected ion monitoring mode
2.2 定量和样品检出限
8个毒杀芬同类物的定量采用单个离子的峰面积。毒杀芬同类物的系列校正标准溶液的质量浓度分别为 0.5、5、25、100、200、300 和 500 μg/L。以PCB204为内标,内标法的校正曲线的线性相关系数均大于0.99。这里,毒杀芬同类物和总毒杀芬的定量都使用平均相对响应因子为一系列校正标准溶液中相对响应因子(RRF)的平均值。只有这些RRF的RSD小于20%才能得到一个合格的,使用定量才可以得到准确的结果。从表1可以看出,所有RRF的RSD均小于20%。此外,8个毒杀芬同类物的有很大的差别,其中P62的比其他同类物的小1到2个数量级。这反映了毒杀芬同类物由负化学电离源产生响应的差别,也是用GC/NCI-MS分析毒杀芬的不足之处。
对于最低的质量浓度0.5 μg/L,8个毒杀芬同类物中只有P62不能被检测到。因而,P62的相对响应因子从5 μg/L开始计算。

式中:Cstd、CI分别为目标化合物和内标的质量浓度(μg/L);Astd、AI分别为目标化合物和内标定量离子的峰面积。所得的Cstd用于计算与真值的相对误差(RE/%)。8个毒杀芬同类物所有各级标准溶液的RE均在±30%之内。因此,可以用在校正标准溶液的浓度范围内定量8个毒杀芬同类物,由此,7个毒杀芬同类物的最低定量限(LLOQ)为0.5 μg/L,P62为5 μg/L。进而,样品检出限(DL)由式(2)求出:

式中:Vex为样品萃取液最后的定容体积(本研究为0.5 mL)、Ws为样品的质量(可为干重、湿重、脂肪(Lipid)质量)或液体样品的体积。将本方法的LLOQ与文献中同位素稀释-气相色谱-串联质谱(ID-GC-MS/MS)的仪器检出限进行了比较。IDGC-MS/MS方法中P26、P50和P62的仪器检出限分别为 3.0、3.0 和 6.0 pg[27]。本方法(进样 1 μL)的LLOQ所对应的仪器检出限分别为0.5、0.5和5.0 pg。可以看出,本方法中P26和P50的仪器检出限比ID-GC-MS/MS方法低很多,而P62则相当。
毒杀芬总量的定量采用可检测到的毒杀芬同类物峰面积的和。具体地说是加合六氯到十氯同类物,即离子 m/z 309、343、379、413 和 447 的峰面积,积分的时间窗口以Hx-Sed为起始至PCB209出峰前(本研究为28~51 min,见图3)。在积分时间窗口内,要对每一个选择离子色谱图中的每一个信噪比大于3(S/N≥3)的色谱峰进行识别,积分出毒杀芬同类物的峰面积,然后加合起来。显然,这是一项相当费时的过程。过去,人们简单地积分色谱基线以上所有峰的面积(area under the curve)来快速得出总面积[9]。由于有干扰物的共流出,这一方法产生正偏差的几率较大。GC/NCI-MS的选择离子扫描模式可以有效地避免这一状况的发生。
对总毒杀芬的定量试验测试了质量浓度为25、50、100、150、200、250 和 500 μg/L 的系列校正标准溶液。以PCB204为内标,除25 μg/L外代回各级标准溶液得到的RE在±30%之内,因此确定总毒杀芬的LLOQ为50 μg/L,并且以此浓度为系列校正标准溶液的最低浓度。

图3 工业毒杀芬校正标准溶液(250 μg/L)在选择离子检测模式下的GC/NCI-MS谱图Fig.3 GC/NCI-MS chromatograms of the technical toxaphene calibration standard(250 μg/L)under the selected ion monitoring mode
2.3 PCB氧反应的监测
由于存在PCB氧反应的缘故,毒杀芬的分析必须要同步检测GC/NCI-MS仪器系统的氧反应水平。PCB氧反应水平(OR)通常用PCB204来测量,由式(3)求出:

式中:A411、A430分别为 PCB204的离子 m/z 411和430的峰面积。PCB氧反应所产生的干扰离子的相对丰度与毒杀芬的不同。以P50和PCB204为例(见图4),P50的定量离子m/z 413是其3个特征离子中丰度最高的,而由PCB204氧反应产生的这3个离子的丰度呈下降的态势。其他含不同Cl原子的毒杀芬和PCB同类物系列有着相似的特点。这非常有利于识别PCB氧反应产生的干扰峰。
一个正常运转且干净的NCI-MS离子源起初可以达到0.2%的氧反应水平,但其会随着分析样品数量的增加而增加。PCB氧反应水平应尽量控制在小于0.5%,不能大于1%。在分析毒杀芬期间,每天吹扫一次离子源可以有效地降低氧反应水平。理论上可以有127个五氯到九氯的PCB单体会由于氧反应而产生干扰,特别是12个PCB八氯单体(PCBs 194~205)会对八氯和九氯的毒杀芬同类物产生干扰。如果样品中PCB的含量很高,即使有低的氧反应水平也会产生大的干扰,因此更要仔细进行色谱峰的逐一识别。具体方法详见文献[22]。

图4 PCB204氧反应产生的干扰和P50在选择离子检测模式下的GC/NCI-MS质谱图Fig.4 GC/NCI-MS selected ion monitoring mass spectra of PCB204 interference due to oxygen reaction and P50
2.4 准确性和精确性
沉积物和鱼样在ASE上由二氯甲烷萃取,经样品净化,替代物DBOFB的回收率在60%~130%,PCB209的回收率在70%~130%。萃取并测定了毒杀芬同类物加标(80 ng/g干重)的沉积物样品,日间重复样的RSD范围是5.4%~12.8%(n=10)。日间测定的同类物平均回收率是(90.8±17.4)%(n=10),说明本方法的准确性和精确性都很高。对于鱼肉样品,笔者已经报道了用美国标准局的鱼标准样品 SM1946进行的确证[22]。谢原利等[26]考察了温度和压力对ASE萃取土壤样品的影响,在温度为70~130℃范围内和8.26~13.78 MPa下,毒杀芬的回收率达90%~110%。本方法所用的ASE萃取条件为100℃和10.3 MPa,也恰好在上述温度和压力的范围内。
2.5 实际样品检测
美国加州蒂华纳河口沉积物中毒杀芬含量见表3,其中,TJR6012的数值是2个平行样的平均值和标准偏差。以5 g沉积物干重为基准,P62的LLOQ是0.5 ng/g,其他7个毒杀芬同类物的 LLOQ是0.05 ng/g。总毒杀芬的LLOQ是5 ng/g。所有8个毒杀芬同类物都被检出,但含量很低,只有个别样品的Hx-Sed,Hp-Sed和P40高于LLOQ。毒杀芬总含量则低于检出限。有报道指出在美国南加州毒杀芬的使用量很小[36],表3的数据表明这一地区基本没有毒杀芬的污染。

表3 蒂华纳河口沉积物中总毒杀芬和8个毒杀芬同类物的含量Table 3 Contents of total toxaphene and the eight congeners in the sediments of Tijuana River estuary ng/g(dry weight)
三文鱼样品中毒杀芬含量见表4,其中的数据是2个平行样的平均值和标准偏差。三文鱼样品的含水量为74%,脂肪含量为33.3%干鱼肉重。以2.5 g干鱼肉重为基准,P62的LLOQ是1.0 ng/g,其他7个毒杀芬同类物的LLOQ是0.1 ng/g,总毒杀芬的LLOQ是10 ng/g。若以鱼肉的湿重为基准,LLOQ分别是0.26、0.026和2.6 ng/g。若以2.5 g干鱼肉中的0.83 g脂肪重为基准,LLOQ分别是3、0.3和30 ng/g。所有8个毒杀芬同类物都被检出,除Hx-Sed外,都高于LLOQ。毒杀芬总含量也高于LLOQ。

表4 三文鱼样品中总毒杀芬和8个毒杀芬同类物的含量(n=2)Table 4 Contents of toxaphene and the eight congeners in the salmon fish tissue(n=2)
3 结论
本研究建立了GC/NCI-MS分析毒杀芬总量和8个毒杀芬同类物的方法。该方法选择性高,检出限低。通过对沉积物和鱼肉样品的分析,证实该方法适用于环境样品和鱼肉样品的分析,也可作为其他领域分析毒杀芬的参考。本研究所用的样品制备方法可以通用于多种疏水性有机污染物的分析,是一种较为经济的多残留方法。随着GC/NCI-MS仪器的普及,该方法可作为毒杀芬的新一代日常检测方法使用。
[1]Voldner E C,Li Y F,Chemosphere,1993,27(10):2073
[2]de Geus H J,Besselink H,Brouwer A,et al.Environ Health Perspect,1999,107:115
[3]Wong F,Alegria H A,Bidleman T F.Environ Pollut,2010,158(3):749
[4]Kucklick J R,Helm P A.Anal Bioanal Chem,2006,386(4):819
[5]Korytar P,van Stee L L P,Leonards P E G,et al.J Chromatogr A,2003,994(1/2):179
[6]Vetter W.Chemosphere,1993,26(6):1079
[7]Maruya K A,Francendese L,Manning R O.Estuaries,2005,28(5):786
[8]Krock B,Vetter W,Luckas B.Chemosphere,1997,35(7):1519
[9]Maruya K A,Walters T L,Manning R O.Estuaries,2001,24(4):585
[10]Smalling K L,Maruya K A.J Sep Sci,2001,24(2):104
[11]Bordajandi L R,Ramos J J,Sanz J,et al.J Chromatogr A,2008,1186(1/2):312
[12]de Geus H J,Baycan-Keller R,Oehme M,et al.J High Resolut Chromatogr,1998,21(1):39
[13]Zhang B,Zheng M H,Liu G R,et al.Chinese Journal of Analytical Chemistry(张兵,郑明辉,刘国瑞,等.分析化学),2012,40(8):1213
[14]Swackhamer D L,Charles M J,Hites R A.Anal Chem,1987,59(6):913
[15]Xia X Y,Crimmins B S,Hopke P K,et al.Anal Bioanal Chem,2009,395(2):457
[16]Skopp S,Oehme M,Chu F L,et al.Environ Sci Technol,2002,36(12):2729
[17]Gouteux B,Lebeuf M,Trottier S,et al.Chemosphere,2002,49(2):183
[18]Chan H M,Yeboah F.Chemosphere,2000,41(4):507
[19]Veyrand B,Venisseau A,Marchand P,et al.J Chromatogr B,2008,865(1/2):121
[20]Fowler B.Chemosphere,2000,41(4):487
[21]Santos F J,Galceran M T,Caixach J,et al.Rapid Commun Mass Spectrom,1997,11(4):341
[22]Lao W J,Tsukada D,Maruya K A.J Chromatogr A,2012,1270:262
[23]SN 0502-95
[24]YC/T 180-2004
[25]Wang M T,Liu Z Y,Mu J,et al.Dyeing and Finishing(王明泰,刘志研,牟峻,等.印染),2006(6):37
[26]Xie Y L,Rao Z,Wang X H,et al.Journal of Instrumental Analysis(谢原利,饶竹,王晓华,等.分析测试学报),2009,28(7):804
[27]Zhang B,Wu J J,Liu G R,et al.Chinese Journal of Chromatography(张兵,吴嘉嘉,刘国瑞,等.色谱),2010,28(5):456
[28]Tian S Q,Mao X H,Miao S,et al.Chinese Journal of Chromatography(田绍琼,毛秀红,苗水,等.色谱),2012,30(1):14
[29]Xie Y L,Rao Z,Wang M,et al.Rock and Mineral Analysis(谢原利,饶竹,王沫,等.岩矿测试),2008,27(5):363
[30]Liu J S,Liu H Y,Zhang H,et al.Chinese Journal of Environmental Science and Technology(刘婕丝,刘红玉,张慧,等.环境科学与技术),2007,30(10):90
[31]Carlin F J,Revells H L,Reed D L.Chemosphere,2000,41(4):481
[32]USEPA.Method 8276-2012.[2013-06-10].http://www.epa.gov/osw/hazard/testmethods/pdfs/8276.pdf
[33]Meng X Z,Blasius M E,Gossett R W,et al.Environ Pollut,2009,157:2731
[34]Lao W,Tsukada D,Greensteint D J,et al.Environ Toxicol Chem,2010,29(4):843
[35]Maruya K A,Wakeham S G,Vetter W,et al.Environ Toxicol Chem,2000,19(9):2198
[36]Li Y F.J Geophys Res-Atmosphere,2001,106(D16):17919
