固相萃取-超高效液相色谱-电喷雾串联质谱法同时检测尿样中的麻黄碱和N-甲基麻黄碱
2013-10-22张福成王朝虹
张 琳,张福成,王朝虹,蒋 晔*,许 萌,李 虹
(1.河北医科大学药学院,河北 石家庄 050017;2.空军总医院,北京 100142;
3.最高人民检察院司法鉴定中心,北京 100040;4.云南省公安厅刑警总队,云南 昆明 650000)
麻黄碱和N-甲基麻黄碱具有扩张支气管、抗过 敏和镇咳作用,是感冒药中的常见成分。其中,麻黄碱具有较强的兴奋作用[1],是国际奥委会严格禁止的兴奋剂。因此,体育赛事前确定运动员是否摄取该类药物时,常需进行尿样等生物检材的检测。对于具有器质性心脏病的人员,服用含麻黄碱的制剂还可引起意外心律紊乱而致死。此外,二者还是制造苯丙胺类毒品的主要原料,从吸毒人员体内采集的血样、尿样及毛发等生物检材中检测该类物质是刑侦机构立案的重要依据。因此,建立快速、灵敏的生物样品中痕量麻黄碱分析方法,可大大延长生物检材的检测周期,用于追溯特殊人群的用药史,对于药物的安全使用具有重要意义。
生物检材成分复杂而待测物含量往往较低,因此常需对生物样品中的待测物进行提取、纯化和富集,以提高检测灵敏度。已报道的生物样品前处理方法有稀释法[2]、蛋白沉淀法[3]、液相萃取法[4-11]、中空纤维液相微萃取法[12]、衍生化法[13-15]、超滤法[16]等。但这些方法均存在一些缺点:稀释法严重污染色谱系统;蛋白沉淀法会导致样品稀释,检测灵敏度降低;液相萃取操作繁琐;衍生化法往往受到衍生化效率的限制;中空纤维微液相萃取法的重现性较差;而超滤法存在浓差极化现象,导致测定结果失真。此外,文献[17-19]报道了用固相萃取-液相色谱-质联用的方法检测生物样品中的麻黄碱,检测灵敏度为0.1~1μg/L,对未及时取样且浓度极低的样品往往无法获得满意的分析结果。因此,本文建立了固相萃取-超高效液相色谱-电喷雾串联质谱法(SPE-UPLC-ESI MS/MS)联用技术同时定量测定尿样中的痕量麻黄碱和N-甲基麻黄碱。该方法能有效提取、纯化和富集待测物,灵敏度高,结果准确。
1 实验部分
1.1 仪器与试剂
ACQUITY UPLC超高相液相色谱仪,Xevo TQ-MS串联四极杆质谱仪和MasslynxTM工作站(美国Waters公司)。Milli-Q超纯水制造系统(美国Millipore公司);固相萃取仪(美国 Waters公司),Oasis HLB和Oasis MCX固相萃取柱(1mL,30mg;美国 Waters公司)。
盐酸麻黄碱(批号:171241- 200404)和盐酸甲基麻黄碱(批号:171247- 200301)对照品购于中国药品生物制品检定所,纯度均大于99.5%;甲醇、乙腈、甲酸均为色谱纯,水为超纯水,其他试剂均为分析纯。
1.2 尿样前处理
尿样采自健康受试志愿者。精密吸取4mL尿液置于离心管中,加入200μL 5mol/L盐酸溶液酸化,在离心力8000g条件下离心5min。取上清液过MCX固相萃取柱(预先分别用1mL甲醇和1 mL水活化、平衡),依次用1mL 0.1mol/L盐酸溶液、1mL甲醇和1mL 5%(体积百分数,下同)氨水溶液淋洗,用1mL甲醇-氨水(95∶5,v/v)溶液洗脱,接收洗脱液,待UPLC-ESI MS/MS分析。
1.3 色谱与质谱条件
ACQUITY UPLC BEH Phenyl色谱柱(10cm×2.1mm,1.7μm);流动相 A为乙腈,流动相B为0.3%甲酸溶液。洗脱梯度:0~3min,流动相A比例由10%升至55%,4min时升至90%,4.1min降至10%,保持1.9min。流速:0.4mL/min;柱温:35℃;样品室温度:10℃;进样体积:2μL。
电喷雾离子源正离子扫描,多反应监测模式;毛细管电压:3.0kV;离子源温度:300℃;去溶剂温度:500℃,去溶剂气流量:1000L/h;锥孔气流量:30L/h;碰撞气流量:0.15mL/min。待测物的监测离子及驻留时间、锥孔电压、碰撞电压等参数见表1。

表1 麻黄碱和N-甲基麻黄碱的质谱参数Table 1 MS/MS parameters of ephedrine and N-methylephedrine
2 结果与讨论
2.1 系统适用性试验
在1.3节的条件下,混合对照品溶液、空白尿样和口服急支糖浆制剂7d后的尿样分别经Oasis MCX固相萃取后的UPLC-MS/MS谱图见图1。空白尿样中不含待测物,且空白基质对待测物的测定无干扰。
2.2 色谱条件的选择
2.2.1 色谱柱的选择
近年来,采用离子对色谱法分析检测生物碱的报道较多[17],但这给质谱检测带来困难。本文在酸性流动相(流动相B为0.1%甲酸溶液)条件下,分别考察了 HSS SB(5cm)、BEH C18(10cm)、CSH C18(5cm)、BEH Phenyl(10cm)4种色谱柱(柱内径均为2.1mm,填料粒径均为1.7μm)对待测物灵敏度和分离度的影响(见图2)。结果表明,以BEH Phenyl色谱柱为分离柱时,待测物的灵敏度较高,且分离度较好。
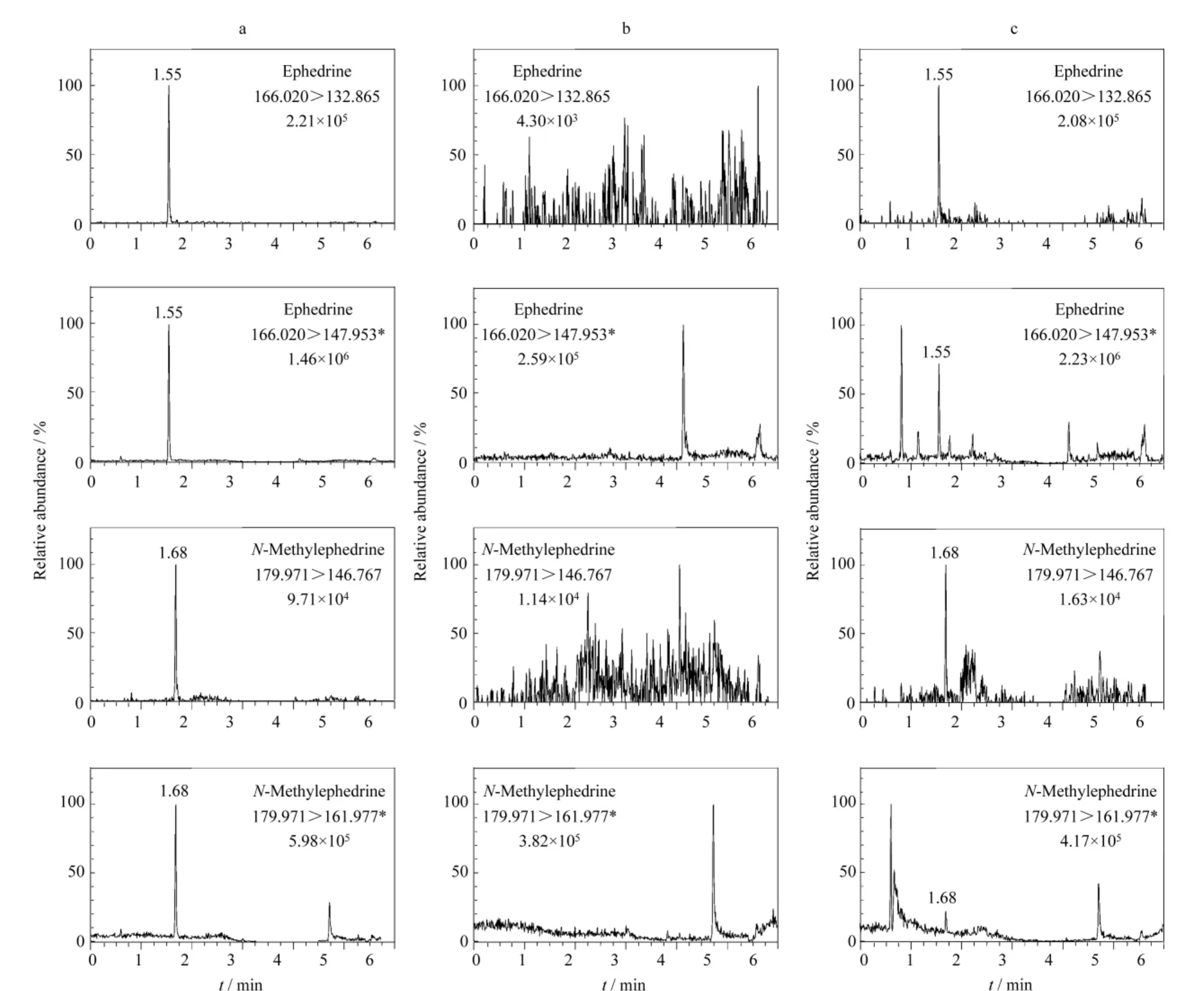
图1 (a)混合对照品溶液(1μg/L)、(b)空白尿样和(c)口服急支糖浆制剂7 d后的尿样分别经MCX柱固相萃取后的UPLC-MS/MS定性和定量离子流谱图Fig.1 UPLC-MS/MS qualitative and quantitative ion current chromatograms of(a)mixed standard solution(1μg/L),(b)human blank urine,and(c)urine after 7 d administering Jizhi Syrup extracted by MCX solid phase extraction*quantitative ion.

图2 不同色谱柱对待测物灵敏度的影响Fig.2 Effect of different chromatographic columns on the sensitivity of the analytes
2.2.2 流动相的选择
以BEH Phenyl色谱柱作为分离柱,分别考察了酸性流动相(流动相B为0.1%甲酸溶液)和碱性流动相(流动相B为10mmol/L碳酸氢铵溶液,pH 10)对待测物灵敏度及峰形的影响(见图3)。结果表明,待测物在酸性流动相下的灵敏度略高于碱性流动相,最终选择酸性流动相。

图3 不同流动相对待测物灵敏度的影响Fig.3 Effect of different mobile phases on the sensitivity of analytes

图4 不同甲酸浓度对待测物灵敏度的影响Fig.4 Effect of different volume percentages of formic acid on the sensitivity of analytes
2.2.3 甲酸浓度的选择
酸性流动相中甲酸的浓度不同,对待测物灵敏度及峰形有不同程度的影响,因此以BEH Phenyl色谱柱作为分离柱,分别考察了0.1%甲酸溶液和0.3%甲酸溶液作为流动相对待测物灵敏度的影响(见图4)。结果表明,以0.3%甲酸水溶液作为流动相时,待测物的灵敏度较高,且峰形较好,最终选择在酸性流动相中添加0.3%的甲酸。
2.3 样品前处理条件的选择
2.3.1 固相萃取柱的选择
由于待测组分呈碱性,在反相色谱填料上的保留很弱,因此很难利用一般的固相萃取小柱达到萃取的目的。本文考察了含有吸附性填料的Oasis HLB固相萃取柱和兼具离子交换和反相保留的Oasis MCX固相萃取柱对样品的提取净化效果。在不含待测物的空白尿中分别添加0.0250、0.250和2.50μg/L的混合标准溶液,按1.2节的方法处理样品,在1.3节的条件下进行测定,比较样品的基质效应(见表2)。
结果表明,HLB柱作为萃取柱会产生严重的离子抑制效应,影响检测灵敏度和准确度,故选择MCX柱作为固相萃取柱。
2.3.2 洗脱条件的选择

表2 Oasis HLB与Oasis MCX固相萃取柱萃取效果的比较(n=6)Table 2 Comparison of the extraction efficiencies of Oasis HLB and Oasis MCX SPE columns(n=6)
为了使待测物在固相萃取柱上得到最好的保留,上样前需要对样品进行酸化以保证待测组分的完全离子化。试验表明,4mL尿液中加入200μL 5 mol/L的盐酸溶液可保证待测组分完全吸附在填料上。尿液等生物样品基质非常复杂,采用质谱检测时基质效应对检测结果的准确性影响很大。为了尽可能地去除基质,本实验采用酸洗、醇洗和碱洗三步清洗,选择性除去非极性的中性物质、酸性以及极性碱性干扰物,最后采用碱性的甲醇溶液进行洗脱。考察了不同比例的甲醇-氨水溶液的洗脱能力。结果表明,以甲醇-氨水(95∶5,v/v)溶液作为洗脱液时,待测物的回收率最高,故选择其作为洗脱液。
2.4 线性范围和检出限
配制麻黄碱和N-甲基麻黄碱质量浓度为1、2、5、10、20、50、100μg/L的混合对照品溶液和内标氘代苯丙胺质量浓度为10μg/L的对照品溶液。吸取上述系列浓度对照品溶液各250μL至7个离心管中,分别加入250μL内标溶液,最后用空白尿稀释至10mL,配制成质量浓度分别为0.0250、0.0500、0.125、0.250、0.500、1.25、2.50μg/L 的样品溶液。按1.2节的方法处理样品,按1.3节的条件进样测定,记录峰面积。以待测物与内标峰面积比(Y)对质量浓度(C,μg/L)进行线性回归,得到麻黄碱和N-甲基麻黄碱在0.0250~2.50μg/L质量浓度范围内的线性方程分别为Y=8.12C-0.143(r=0.9998)和 Y =4.09C+0.735(r=0.9992)。取线性最低点溶液逐倍稀释,按1.3节的条件进样测定,得麻黄碱和N-甲基麻黄碱的检出限 均 为 0.01 μg/L (S/N = 3),均高于文献[5-7,12,14-17,19]报道。
2.5 精密度、准确度、基质效应及提取回收率
配制麻黄碱和N-甲基麻黄碱低、中、高3个浓度分别为0.0250、0.250和2.50μg/L的质控(QC)样品,每个浓度6份,按1.2节的方法处理样品,在1.3节的条件下进行测定,记录峰面积As,计算日内精密度和准确度;连续测定3d,计算日间精密度。同时,取空白尿,按1.2节的方法处理后,配制成低、中、高3个浓度的溶液,每个浓度6份,在1.3节的条件下进行测定,记录峰面积Am;另取上述3个浓度的对照品溶液,不经萃取直接进样,记录峰面积为Astd。以As/Am计算提取回收率,Am/Astd计算基质效应,结果见表3。

表3 麻黄碱和N-甲基麻黄碱的精密度、准确度、基质效应及提取回收率(n=6)Table 3 Precisions,accuracies,matrix effects and recoveries of ephedrine and N-methylephdrine(n=6)
2.6 样品稳定性
配制麻黄碱和N-甲基麻黄碱低、中、高3个质量浓度分别为0.0250、0.250和2.50μg/L的质控样品,每个浓度3份。将上述样品于室温放置24h及经过3次冻融循环后,按1.2节方法处理,并在1.3节的条件下进行测定,考察其短期稳定性及冻融稳定性(见表4)。结果表明,麻黄碱和N-甲基麻黄碱在上述条件下均稳定。

表4 麻黄碱和N-甲基麻黄碱的稳定性(n=3)Table 4 Stabilities of ephedrine and N-methylephedrine(n=3)
2.7 真实样品测定
采集健康志愿者口服10mL急支糖浆制剂7d后的尿样,按1.2节方法处理样品,并按1.3节的条件进行测定,尿样中麻黄碱和N-甲基麻黄碱的质量浓度分别为0.034和0.137μg/L。表明该方法能够有效测定尿样中痕量的麻黄碱和N-甲基麻黄碱,大大提高检测灵敏度,并延长了尿样中麻黄碱和N-甲基麻黄碱的检测周期。
3 结论
本实验采用Oasis MCX固相萃取柱对尿样进行提取、纯化和富集,建立了SPE-UPLC-ESI MS/MS方法分析尿样中痕量麻黄碱和N-甲基麻黄碱,显著提高了检测灵敏度,大大延长了尿样中麻黄碱和N-甲基麻黄碱的检测周期,为未及时取样且浓度极低的样品中痕量麻黄碱和N-甲基麻黄碱的检测提供了有效的方法。
[1]Abourashed E A,El-Alfy A T,Khan I A,et al.Phytother Res,2003,17:703
[2]Li K,Chen J,Xu X,et al.Chinese Journal of Pharmaceuticals(李坤,陈钧,徐新,等.中国医药工业杂志),2009,40(9):695
[3]Feng Y,Huang Y,Lai W L,et al.Journal of Guangdong College of Pharmacy(冯怡,黄羽,赖伟玲,等.广东药学院学报),2008,24(6):572
[4]Ren L,Deng X X,Bi K S,et al.Chinese Journal of Pharmaceutical Analysis(任磊,邓新秀,毕开顺,等.药物分析杂志),2007,27(10):1544
[5]Liu Q Y,Zhang Z H,Chen B,et al.Journal of Instrumental Analysis(刘庆艳,张朝辉,陈波,等.分析测试学报),2008,27(1):26
[6]Li S G,Li H Y,Cai Z,et al.Chinese Journal of Analytical Chemistry(李斯光,李海燕,蔡卓,等.分析化学),2009,37(8):1137
[7]Wang L L,Fang Y,Ge W H.China Pharmacy(王璐璐,方芸,葛卫红.中国药房),2007,18(32):2510
[8]Hu Z,Zou Q,Tian J,et al.J Chromatogr B,2011,879:3937
[9]Wang P Y,Zhang H M,Wang C F,et al.Chinese Journal of Information on TCM(王朋义,张海鸣,王成芳,等.中国中医药信息杂志),2012,19(1):56
[10]Zheng Z,Yan T,Chen W,et al.Xenobiotica,2012,42(8):775
[11]Salam R A A,Hadad G M,Hameed E A A.J Liq Chromatogr Relat Technol,2013,36:384
[12]Chen X,Bai X H,Wang X,et al.Chinese Journal of Chromatography(陈璇,白小红,王晓,等.色谱),2010,28(12):1144
[13]LeBelle M J,Savard C,Dawson B A,et al.Forensic Science International,1995,71:215
[14]Herráez-Hernández R,Campíns-FalcóP.J Chromatogr A,2000,893:69
[15]Aymard G,Labarthe B,Warot D,et al.J Chromatogr B,2000,744:25
[16]Sorensen L K.J Chromatogr B,2011,879:727
[17]Chen X H,Li X P,Yao X P.Chinese Journal of Health Laboratory Technology(陈晓红,李小平,姚浔平.中国卫生检验杂志),2006,16(6):691
[18]He Y N,Hu Y Q,Yang H Y,et al.China Pharmacy Journal(何颖娜,胡玉钦,杨汉煜,等.中国药学杂志),2009,44(6):464
[19]Chen X H,Qiu P H,Jin M C et al.Physical Testing and Chemical Analysis Part B:Chemical Analysis(陈晓红,仇佩虹,金米聪,等.理化检验:化学分册),2006,42(10):790
