反相高效液相色谱法同时测定甘草酸单铵盐原料药主成分及有关物质含量
2013-10-22赵燕燕刘丽艳韩媛媛李月秋石敏健
赵燕燕,刘丽艳,韩媛媛,李月秋,王 艳,石敏健
(1.河北大学医学实验中心,河北 保定 071000;2.河北大学化学与环境科学学院,河北省分析科学技术重点实验室,河北省化学生物学重点实验室,河北 保定 071002)
甘草酸单铵盐1992年被国家卫生部定为治疗慢性肝炎的首选药物,其原料药主成分是18α-甘草酸(18α-glycyrrhizic acid,18α-Gly)和18β-甘草酸(18β-glycyrrhizic acid,18β-Gly),以及2个主要的有关物质,分别命名为有关物质A和有关物质B。18α-Gly和18β-Gly为一对差向异构体,其空间构型的微小变化使两者在理化性质、药效学、药代动力学及不良反应等方面存在着显著差异。目前关于甘草酸制剂检测方法的研究报道很多,如高效液相色谱-质谱联用法(LC-MS)[1,2]、毛细管区带电泳法[3,4]、高效液相色谱法[5-10]、气相色谱法[11]、薄层色谱法[12]、极谱催化波法[13]、紫外分光光度法[14]等。高效液相色谱法具有准确、快捷、灵敏度高、重现性好和应用广泛等优点,是原料药和制剂质量标准研究中常用的方法。原料药中18α-Gly、18β-Gly所占比例及有关物质含量是控制其制剂质量和安全的重要指标。在中国药典2010版中没有收载甘草酸单铵盐质量标准。在欧洲药典7.0版(EP7.0)和英国药典2012版(BP2012)收载的甘草酸单铵盐质量标准中,将其主成分结构归属为18β-Gly,将其中2个有关物质分别归属为有关物质A(相对保留时间约为0.8)和18α-Gly(相对保留时间约为1.2),并未提及有关物质 B。经查阅相关文献[5],EP7.0、BP2012和中国国家药品标准WS1-XG-2002收载的甘草酸单铵盐质量标准中的流动相为酸性体系,对18α-Gly、18β-Gly异构体没有分离选择性,无法同时对其主成分18α-Gly、18β-Gly的含量及有关物质进行准确定量分析。由此,对主成分异构体的分离以及对色谱图中相对保留时间约为1.2的物质的结构归属有待商榷。现有的甘草酸类制剂标准中尚未标注18α-Gly、18β-Gly所占比例,造成产品内在质量控制的缺失。目前对甘草酸类制剂中差向异构体的分析已有相关报道[15,16],但有关甘草酸单铵盐原料药主成分及有关物质含量检测方法的研究尚未见文献报道。本实验在参考国内外相关文献和各国药典的基础上,建立了采用反相高效液相色谱(RP-HPLC)同时测定甘草酸单铵盐原料药主成分及有关物质含量的方法,可用于甘草酸单铵盐原料药主成分含量及有关物质的检测分析。
1 实验部分
1.1 仪器、试剂与材料
LC-10ATVP高效液相色谱仪(日本岛津公司),包括SPD-M10Avp二极管阵列检测器(DAD)和CLASS-VP工作站;UV-2550紫外分光光度计(日本岛津公司);AUW120D十万分之一电子分析天平(日本岛津公司);DELTA-320型酸度计(梅特勒-托利多仪器有限公司);超纯化水机(重庆颐洋企业发展有限公司)。
甘草酸单铵盐对照品(中国食品药品检定研究院,批号:Y0 000433)、18α-Gly对照品(ALPS制药,批号:10B001S)、18β-Gly对照 品 (ALPS 制药,批号:05C11S)、有关物质 A对照品(ALPS制药,批号:10B002S)、有关物质B对照品(ALPS制药,批号:10B003S),以上对照品纯度均高于99%;甘草酸单铵盐原料药(A厂,批号:V150 860001;B厂,批号:06802N130;C厂,批号:11C8、10E4;D 厂,批号:09 120061)。高氯酸铵、氨水、高氯酸、冰醋酸、三乙胺、磷酸均为分析纯。甲醇、乙腈为色谱纯(天津市康科德科技有限公司)。水为二次去离子水。
1.2 溶液的配制
对照品溶液:精密称定各对照品适量,分别置于100mL容量瓶中,加入50%甲醇水溶液溶解并定容,分别制成质量浓度为1000mg/L的对照品储备液,于4℃冰箱中保存。精密量取5mL对照品储备液,置于100mL容量瓶中,用流动相定容,摇匀,作为对照品溶液。
供试品溶液:精密称定各原料药适量,分别置于100mL容量瓶中,加入50%甲醇水溶液溶解并定容,分别制成质量浓度为1000mg/L的供试品储备液,于4℃冰箱中保存。精密量取5mL供试品储备液,置于100mL容量瓶中,用流动相定容,摇匀,作为供试品溶液。
混合对照品溶液:分别精密量取18α-Gly、18β-Gly、有关物质A和有关物质B对照品储备液适量,置于同一容量瓶中,用流动相定容,摇匀,作为混合对照品溶液。
专属性试验测试液:精密量取供试品储备液(C厂,批号:11C8)5mL,加入6mol/L NaOH 溶液1 mL,常温下放置24h,用6mol/L HCl溶液调pH值至中性,用流动相稀释至10mL,摇匀,作为碱破坏样品溶液。精密量取供试品储备液(C厂,批号:11C8)5mL,加入6mol/L HCl溶液1mL,常温下放置24h,用6mol/L NaOH溶液调pH值至中性,用流动相稀释至10mL,摇匀,作为酸破坏样品溶液。精密量取供试品储备液(C厂,批号:11C8)5 mL,在160℃恒温箱中加热近干,用流动相稀释至10mL,摇匀,作为高温破坏样品溶液。精密量取供试品储备液(C厂,批号:11C8)5mL,加入质量分数为3%的双氧水5mL,放置24h,用流动相稀释至10mL,摇匀,作为氧化破坏样品溶液。
1.3 色谱条件
色谱柱:Durashell C18柱(250mm×4.6mm,5 μm);流动相:10mmol/L高氯酸铵(氨水调节pH 8.20)-甲醇(48∶52,v/v);流速:0.80mL/min;检测波长:254nm;柱温:50℃;进样量:10μL。
2 结果与讨论
2.1 色谱分离条件的建立
2.1.1 流动相组成、盐浓度及pH值的选择
实验分别考察了反相色谱常用的有机相溶剂(甲醇和乙腈)与水的混合体系为流动相以及在流动相中分别添加0.5%冰醋酸、0.6%冰醋酸、60%高氯酸、0.01mol/L磷酸、0.10mol/L磷酸盐缓冲液等对18α-Gly、18β-Gly、有关物质 A和有关物质B分离效果及其灵敏度的影响。结果表明,在4种待测物混合对照品能够达到基线分离的前提下,以甲醇-水-60%高氯酸(55∶45∶0.5,v/v/v,氨水调pH值至8.00)为流动相,分离效果最好,灵敏度最高,保留时间最短。该流动相虽解决了两异构体分离的问题,但甲醇-水-60%高氯酸体系的pH值在1.7左右,以氨水调pH至8.00的操作比较繁琐;因此采用高氯酸铵代替高氯酸,再用氨水调pH值至8.20,色谱行为不变,且操作简单、快捷。
18α-Gly和18β-Gly是一对差向异构体,流动相的pH值显著影响该异构体的拆分,同时分离18α-Gly和18β-Gly需在pH≥7的条件下进行。欧洲药典7.0版、英国药典2012版、中国国家药品标准WS1-XG-2002收载的甘草酸单铵盐药品标准方法中采用的流动相分别为冰醋酸-乙腈-水(6∶380∶614,v/v/v)(欧洲药典和英国药典)和0.01mol/L磷酸-乙腈(62∶38,v/v)(WS1-XG-2002),均为酸性体系,对18α-Gly、18β-Gly异构体没有分离选择性(见图1)。
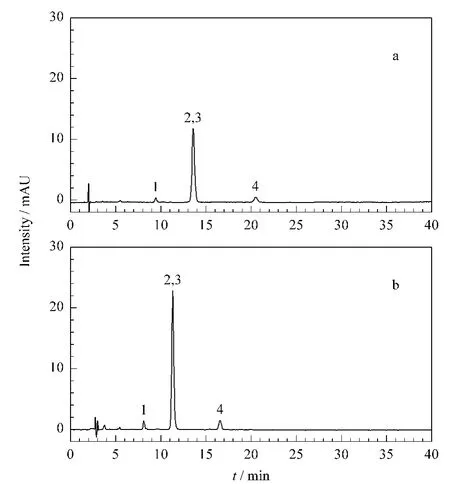
图1 采用(a)欧洲药典7.0版/英国药典2012版和(b)中国国家药品标准收载的甘草酸单铵盐标准方法得到的色谱图Fig.1 HPLC chromatograms of raw material drug of ammonium glycyrrhizinate with the methods of(a)European Pharmacopoeia 7.0 version/British Pharmacopoeia 2012 version and (b)the National Drug Standards of China
实验考察了高氯酸铵浓度在5~100mmol/L范围内对各色谱峰的分离度、灵敏度以及保留时间的影响。结果表明,当高氯酸铵的浓度从5 mmol/L升高至10mmol/L时,4种待测物的灵敏度均达到最大值,但对分离度及各色谱峰保留时间的影响不明显;继续增加高氯酸铵的浓度,可以改善4种待测物的分离度,但会导致灵敏度下降,各色谱峰的保留时间延长。实验选择高氯酸铵浓度为10 mmol/L。
实验考察了流动相pH值(6.45~8.93)对各色谱峰的分离度、灵敏度以及保留时间的影响。结果表明,pH值从6.45升高至8.20,4种待测物的灵敏度均增加,各色谱峰的保留时间明显缩短;pH=8.20时,18α-Gly和18β-Gly刚好达到基线分离;继续增大pH值,除有关物质A外,其他3种待测物的灵敏度仍随之增加,但对18α-Gly、18β-Gly异构体的分离选择性降低。兼顾分离度和灵敏度的要求,实验选择pH值为8.20。
2.1.2 有机相加入比例的选择
实验考察了流动相中甲醇加入比例(45%、50%、52%、55%、60%,v/v)对各色谱峰的分离度、灵敏度以及保留时间的影响。结果表明,随着甲醇加入比例的增加,各色谱峰的保留时间逐渐缩短、灵敏度逐渐增加;当甲醇的加入比例为52%时,差向异构体18α-Gly和18β-Gly恰好达到基线分离;甲醇含量继续增大,两差向异构体的分离度小于1.5,不符合药典要求。实验选用甲醇加入比例为52%。
2.1.3 柱温及流速的选择
许多研究表明,温度对手性化合物的保留因子、分离度和柱效有一定的影响[17]。实验考察了柱温(25、30、40、45、50℃)对各色谱峰的分离度、灵敏度以及保留时间的影响。结果表明,随着柱温的升高,4种待测物的灵敏度增加,各色谱峰的保留时间缩短,分离度降低,但不影响基线分离。当柱温大于50℃时,会降低色谱柱使用寿命。在不影响分离且不损坏色谱柱的基础上,实验选择柱温为50℃。
实验考察了流速(1.20、1.00、0.80、0.60、0.40 mL/min)对各色谱峰的分离度、灵敏度以及保留时间的影响。结果表明,随着流速的降低,理论塔板高度降低,理论塔板数增加,4种待测物的灵敏度和分离度均有一定程度的提高,但各色谱峰的保留时间显著延长;当流速为0.80mL/min时,18α-Gly和18β-Gly达到基线分离;流速继续降低,各色谱峰的保留时间延长,影响快速分离。综合分析,实验选择0.80mL/min为最佳流速。
实验表明,采用1.3节色谱条件时的分离效果最佳。
2.2 系统适用性试验
分别量取混合对照品溶液、供试品溶液、供试品加标溶液按1.3节色谱条件进样分析,系统适用性试验测定结果见表1,色谱图见图2。

表1 系统适用性试验测定结果Table 1 Results of system suitability test
2.3 方法专属性试验
分别量取酸破坏、碱破坏、高温破坏、氧化破坏样品溶液,按1.3节色谱条件进样分析。结果表明,在酸、碱、高温、氧化等破坏条件下得到的降解产物在该色谱条件下均能与18α-Gly、18β-Gly、有关物质A和有关物质B色谱峰良好分离,说明所建立的HPLC法具有良好的专属性。
2.4 方法学考察
2.4.1 线性关系和检出限

图2 (a)混合标准品、(b)供试品及(c)加标供试品的色谱图Fig.2 HPLC chromatograms of(a)a mixture of standards,(b)a sample and(c)the sample spiked with the mixture of standards
精密量取18α-Gly、18β-Gly、有关物质 A 和有关物质B对照品储备液各1mL,分别置于10mL容量瓶中,用流动相稀释至刻度,摇匀,制成质量浓度各为100mg/L的对照品溶液,用流动相逐级稀释,得到一系列质量浓度不同的溶液,按照1.3节色谱条件进样分析,记录色谱图。以峰面积Y对质量浓度X(mg/L)作图,绘制标准曲线;按3倍信噪比(S/N=3)确定检出限。甘草酸单铵盐中主成分及有关物质的线性方程、线性范围及检出限见表2。

表2 甘草酸单铵盐中主成分及有关物质的线性关系及检出限Table 2 Regression relationships and detection limits of principal components and related substances of ammonium glycyrrhizinate
2.4.2 仪器的精密度
取混合对照品溶液,按照1.3节色谱条件进样分析,连续进样6次。结果表明,各色谱峰的保留时间波动小于0.02min(n=6),峰面积的 RSD为0.05%~0.36%(n=6)。
2.4.3 方法的精密度
取同一批次的甘草酸单铵盐原料药(C厂,批号:11C8)的供试品溶液6份,按照1.3节色谱条件进样分析。结果表明,各色谱峰的保留时间波动小于0.02min(n=6),峰面积的 RSD 为0.06%~0.57%(n=6)。
2.4.4 稳定性
取甘草酸单铵盐原料药(C厂,批号:11C8)的供试品溶液,按照1.3节色谱条件,分别放置0、6、12、24、48、72h时进样分析,各色谱峰峰面积的RSD范围为0.39%~1.06%(n=6),表明制备好的供试品溶液在3d内检测稳定性良好。
2.4.5 回收率
精密量取甘草酸单铵盐原料药(C厂,批号:11C8)供试品溶液,按该供试品溶液中各待测物浓度的80%、100%、120%分别加入18α-Gly、18β-Gly、有关物质A和有关物质B的对照品,按照1.3节色谱条件进样分析,记录色谱图。甘草酸单铵盐中主成分及有关物质的加标回收率见表3。

表3 甘草酸单铵盐中主成分及有关物质的加标回收率(n=3)Table 3 Spiked recoveries of principal components and related substances of ammonium glycyrrhizinate(n=3)
2.5 实际样品中主成分及有关物质含量的测定
取对照品溶液,按照1.3节色谱条件进样分析,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的25%。分别取不同厂家生产的甘草酸单铵盐供试品溶液,按照1.3节色谱条件进样分析,记录色谱图至供试品溶液中主峰18β-Gly保留时间的3倍,分别计算4种待测物的含量,结果见表4,甘草酸单铵盐对照品的色谱图见图3。

表4 不同生产厂家的甘草酸单铵盐原料药中主成分及有关物质的含量(n=3)Table 4 Contents of principal components and related substances in raw material drugs of ammonium glycyrrhizinate from different manufacturers(n=3)

图3 甘草酸单铵盐对照品的色谱图Fig.3 HPLC chromatogram of reference substances of ammonium glycyrrhizinate
3 结论
本工作在参考国内外相关文献和各国药典的基础上,建立了同时测定甘草酸单铵盐原料药主成分及有关物质含量的RP-HPLC法。与欧洲药典7.0版、英国药典2012版和中国国家药品标准 WS1-XG-2002收载的甘草酸单铵盐质量标准方法相比,实现了对甘草酸单铵盐原料药主成分异构体18α-Gly和18β-Gly的有效分离,使其主成分以及有关物质A和有关物质B在单一流动相、单流速、等度洗脱条件下取得良好的分离。该方法分离时间短,检出限低,分离度好,线性范围宽,结果准确、可靠,可作为甘草酸单铵盐的检测方法,用于甘草酸单铵盐原料药主成分、有关物质含量的检测与分析,从而用于其原料药和制剂的质量控制,对甘草酸单铵盐质量标准的修订具有一定的参考价值。
[1]Zou Q G,Wei P,Li J,et al.Biomed Chromatogr,2009,23:54
[2]Lin Z P,Qiu S X,Wufuer A,et al.J Chromatogr B,2005,814:201
[3]Sabbioni C,Mandrioli R,Ferranti A,et al.J Chromatogr A,2005,1081:65
[4]Kvasnicka F,Voldrich M,Vyhnalek J.J Chromatogr A,2007,1169:239
[5]Koga K,Ohmachi K,Kawashima S,et al.J Chromatogr B,2000,738:165
[6]Sabbioni C,Ferranti A,Bugamelli F,et al.Phytochem Anal,2006,17:25
[7]Hennell J R,Lee S,Khoo C S,et al.J Pharm Biomed Anal,2008,47(3):494
[8]Yuan X D,Hu M Y,Koh H L,et al.Curr Pharm Anal,2011,7(2):133
[9]Fanali S,Aturki Z,D’Orazio G,et al.J Sep Sci,2005,28(9/10):982
[10]Li H,Lu D Q,Liu W M.Chinese Journal of Chromatography(李晖,卢定强,刘伟民.色谱),2004,22(3):258
[11]LüJ,Wu X M,Zhou Q.Chinese Pharmaceutical Journal(吕坚,吴锡铭,周晴.中国药学杂志),1993,28(9):552
[12]Vampa G,Benvenuti S.J Chromatogr,1991,534:479
[13]Guo W,Zhao M R,Song J F,et al.Chemical Journal of Chinese Universities(过玮,赵明仁,宋俊峰,等.高等学校化学学报),1996,17(10):1541
[14]Zhao H,Lu C H,Xuan C X.Chinese Journal of Hospital Pharmacy(赵鸿,鹿程华,玄春香.中国医院药学杂志),2006,26(12):1577
[15]LüZ Y,LüY G,Che B Q,et al.Chinese Journal of Pharmaceutical Analysis(吕昭云,吕亚光,车宝泉,等.药物分析杂志),2010,30(12):2372
[16]Song H B,Liu Q,Zhao H,et al.Chinese Journal of Pharmaceutical Analysis(宋红波,刘茜,赵辉,等.药物分析杂志),2012,32(5):883
[17]Ma H P,Li L S,Chen H M,et al.Chinese Journal of Analytical Chemistry(马海萍,李来生,陈会明,等.分析化学),2010,38(2):158
