2-苯基苯并咪唑的合成及工艺优化
2013-09-27杨晓峰陈志萍
刘 博,杨晓峰,靳 强,陈志萍
(中北大学理学院化学系,山西 太原 030051)
苯并咪唑及其衍生物具有良好的生物活性,广泛应用于医药、农药、兽药、抗真菌剂等领域[1-4]。苯并咪唑衍生物与金属离子形成配合物后,可用作模拟天然超氧化物歧化酶(SOD)的活性成分[5]以及OLED发光材料[6]。因而苯并咪唑化合物的合成研究受到了国内外学者的广泛关注。
通常情况下该类化合物有2种合成途径[7-8]:一是以邻苯二胺和有机羧酸或衍生物为原料,通过环化、脱水反应来合成。该方法通常需要强酸、高温等条件,反应时间也较长且产率较低;二是以邻苯二胺和醛为原料,在氧化剂存在下进行反应来合成,常见的氧化剂有[9-11]DDQ、MnO2、Pb(OAc)4、过硫酸氢钾、Na2S2O5、氨基磺酸、偏钒酸铵、I2-H2O2等。该方法可以降低反应温度,减少反应时间,较传统方法有了很大的改善,但由于氧化剂的参与,致使副产物增多,给目标化合物的后续分离提纯造成了困难。为了消除固体氧化剂对产物分离的影响,Singh 等[12]对该方法进行了改进,以MeCN或DMF作溶剂,在Fe(Ⅲ)/Fe(Ⅱ)氧化还原体系催化下,直接以氧气为氧化剂,合成苯并咪唑衍生物。Kawashita等[13]则用活性炭负载氧气作氧化剂,合成苯并咪唑衍生物。本研究在上述工作的基础上,对合成工艺进一步改进,以邻苯二胺和苯甲醛为原料,不借助任何催化剂和载体,直接以氧气为氧化剂,进行2-苯基苯并咪唑的合成,并对合成工艺进行了优化。
1 实验部分
1.1 实验药品
邻苯二胺,甲醇,苯甲醛,所有试剂均为分析纯。
1.2 2-苯基苯并咪唑的合成及工艺优化
将10mmol(1.08g)邻苯二胺溶于30mL甲醇,置于100mL四口烧瓶中,搅拌下缓慢滴加10mL溶解有20mmol(2.04ml)苯甲醛的甲醇溶液,水浴升温至45℃,通入氧气作为氧化剂,回流反应3h,TLC跟踪反应进程,减压蒸馏,得黄色糊状固体,95%乙醇重结晶,活性炭脱色,得到目标化合物。
改变反应温度、原料配比、反应时间、溶剂用量等反应条件,考察各因素对目标化合物产率的影响,确定合成反应的最佳工艺条件。
1.3 目标化合物的表征
用X-4显微熔点测试仪测定样品的熔点;采用KBr 压片,FIIR-8400S傅里叶变换红外光谱仪测定样品的红外光谱;将目标化合物溶于DMSO-d6,利用BRUKER-DXR300核磁共振仪测定样品的氢核磁共振图谱。
2 结果与讨论
2.1 2-苯基苯并咪唑的结构分析与表征
2-苯基苯并咪唑的合成路线如Scheme1所示。

(1)熔点:经X-4显微熔点测试仪测试,目标化合物的熔点为293~295℃,与文献一致。
(2)FTIR:图1为2-苯基苯并咪唑(A)和原料邻苯二胺(B)的红外图谱。在曲线A、B中,1635、1593、1504、1458cm-1附近的吸收峰为苯环 C=C骨架伸缩振动;752cm-1附近的吸收峰为苯环1,2-二元取代后C-H 键面外弯曲振动;3030cm-1附近的吸收峰为苯环的C-H伸缩振动;曲线B中在3382、3363cm-1处的吸收峰为伯胺的N-H伸缩振动,曲线A中,在3047cm-1处出现仲胺基
N-H的伸缩振动吸收峰,而在3500~3200cm-1范围内伯胺的N-H伸缩振动吸收峰消失,说明伯胺基已与醛基反应,生成仲胺。进一步对比曲线A、B中N-H伸缩振动峰的位置与峰形可发现曲线A的振动峰位置较之曲线B发生了红移300cm-1且峰形趋于平坦,这是因为目标化合物呈大共轭体系[14],致使N-H伸缩振动力常数减弱,吸收峰发生红移;同时,咪唑环上的氢较为活泼,呈缔合态存在,其峰形减弱。上述N-H伸缩振动峰位置的红移与峰形变弱进一步证明反应生成了苯并咪唑类化合物。曲线A中,在740、702cm-1处出现2个强吸收峰,可归结为单取代苯特征吸收峰,即咪唑环2位取代苯基的C-H面外弯曲振动吸收峰,表明目标化合物中有一取代苯的存在。综上所述,胺基已与醛基反应,生成咪唑。
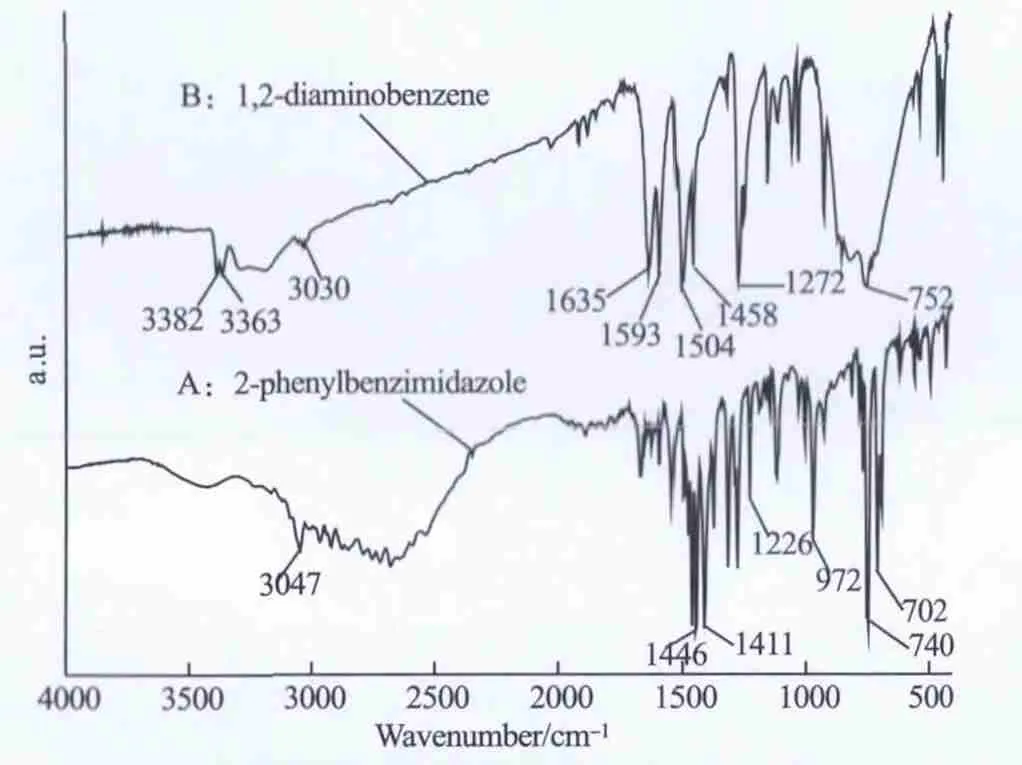
图1 2-苯基苯并咪唑与邻苯二胺红外图谱Fig.1 FTIR spectra of 2-Phenylbenzimidazole and 1,2-diaminobenzene
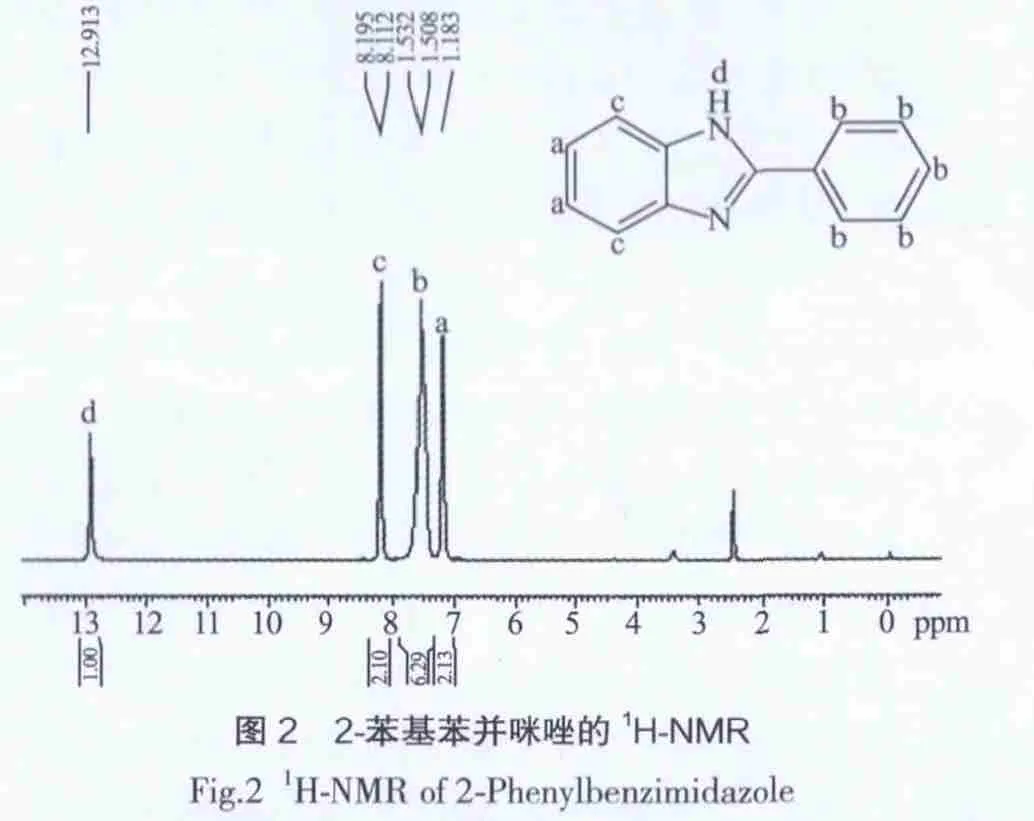
图2 2-苯基苯并咪唑的1H-NMRFig.2 1H-NMR of 2-Phenylbenzimidazole
(3)1HNMR:图2为2-苯基苯并咪唑氢核磁共振谱图,图中出现4组共振峰,积分面积之比(Ha∶Hb∶Hc∶Hd)为 2∶5∶2∶1。 在 δ=12.913(d)处出现的共振峰,可归属仲胺基的1个活泼H,胺基上的H处于它所连接苯环的去屏蔽区,因此,化学位移向低场移动。在苯环区(化学位移δ= 7.0~8.0)有3组共振峰出现,可归属为苯并咪唑苯环的4个H以及单取代苯环的5个H;在δ=7.508~7.532(b)处出现的共振峰,归属为单取代苯环的5个H,由于取代基为不饱和的苯并咪唑基团,与苯环形成共轭,使苯环电子云密度降低,表现为去屏蔽效应,因此,化学位移向低场移动。在δ=8.172~8.195(c)处的共振峰,归属为苯并咪唑苯环的3、6位上的H;在δ=7.183(a)处的峰,归属为苯并咪唑苯环的4、5位上的H。在δ= 2.565处为溶剂DMSO-d6的吸收峰。由氢核磁图谱可进一步证实合成产物为目标化合物2-苯基苯并咪唑。
2.2 合成工艺优化
2.2.1 反应温度选择
固定邻苯二胺的量为10mmol、苯甲醛的量为20mmol、反应时间为3 h、甲醇/邻苯二胺的液固比(mL/g)为37.0(甲醇用量为40mL),考察反应温度对产率的影响,实验结果如图3所示。
图3显示,随着反应温度的升高,2-苯基苯并咪唑产率先增大后减小,反应温度为45℃时,产率最高可达71.2%。这是由于随着温度的升高,首先达到主反应所需的活化能,反应速率增大,产率增大;当温度继续升高时,邻苯二胺有可能被氧气氧化为2,3-二氨基吩嗪[15],吩嗪类化合物一般是在氧化酶[16]催化或高温高压下合成,所需活化能较高,升高温度致使吩嗪类副产物的生成速率增大,使目标化合物产率降低。因此,在此条件下,合成反应较为适宜的温度为45℃。
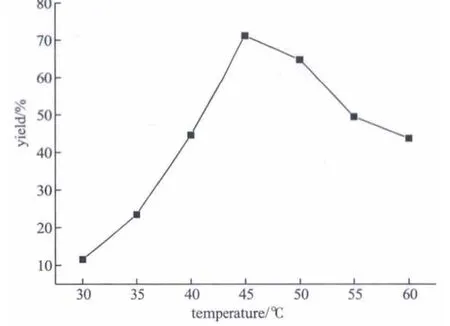
图3 温度对反应产率的影响Fig.3 Influence of temperature on reaction yield
2.2.2 原料配比选择
固定邻苯二胺的量为10mmol、反应时间为3h、温度为45℃、甲醇/邻苯二胺的液固比(mL/g)为37.0(甲醇用量为40mL) (甲醇用量为40mL),改变苯甲醛与邻苯二胺的摩尔比,考察原料配比对产率的影响,结果如图4所示。
由图4可知,随着苯甲醛用量的增加,2-苯基苯并咪唑的产率也是先增大后减小趋势,当原料配比为n(苯甲醛)/n(邻苯二胺)=2,反应产率最高。这可能是由于在氧气存在的条件下,苯甲醛极易被氧化为苯甲酸,苯甲醛不能完全地与胺基反应,因此,此反应需要过量的苯甲醛;当苯甲醛配比用量超过2时,随着苯甲醛的用量增大,生成的苯甲酸等副产物也随之增多,给后续的分离提纯造成了困难,产率降低。因此,在此条件下,较为适宜的原料配比为n(苯甲醛)/n(邻苯二胺)=2。
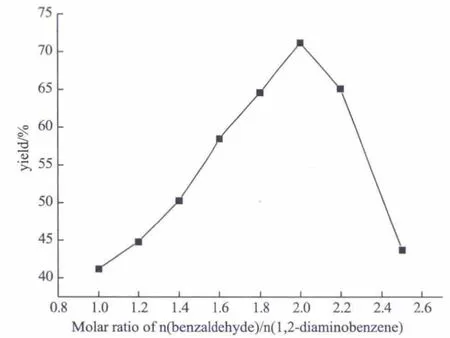
图4 原料配比对反应产率的影响Fig.4 Influence of molar ratio on reaction yield
2.2.3 反应时间的选择
固定邻苯二胺的量为10mmol、苯甲醛的量为20mmol、反应温度为45℃、甲醇/邻苯二胺的液固比(mL/g)为37.0(甲醇用量为40mL) (甲醇用量为40mL),改变反应时间,考察反应时间对产率的影响,实验结果如图5所示。
由图5可知,随着反应时间的延长,2-苯基苯并咪唑的产率也呈先增大后减小趋势,反应3h产率最高。3h产率最高的原因目前尚不清楚,可能与副反应的加剧有关,有待于进一步探索和研究。
2.2.4 溶剂用量的选择
固定邻苯二胺的量为10mmol、苯甲醛的量为20mmol、反应温度为45℃、反应时间为3h,选择适宜的溶剂用量,考察反应体系液固比对产率的影响,实验结果如图6所示。
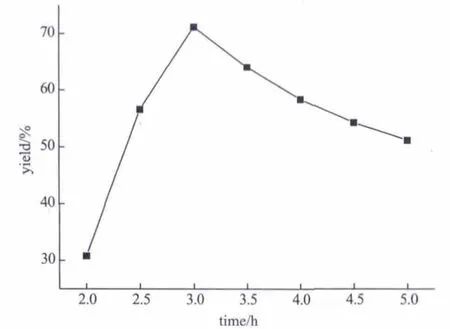
图5 时间对反应产率的影响Fig.5 Influence of time on reaction yield
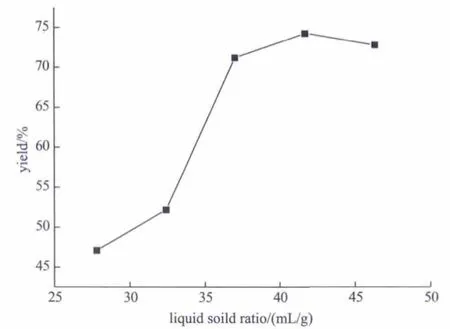
图6 反应体系液固比对反应产率的影响Fig.6 Influence of liquid solid ratio on reaction yield
图6显示,当邻苯二胺用量一定时,2-苯基苯并咪唑产率随溶剂用量的增加而增加,当体系液固比超过37.0后,产率基本保持不变。这是由于体系液固比小于37.0时,溶剂未能将原料充分溶解,反应不完全,产率较低;当体系液固比超过37.0时,溶剂已将原料溶解,可进行充分反应,因此产率变化不大。在此条件下,体系较为适宜的液固比(mL/g)为37.0,此时甲醇用量为40mL。
3 结论
(1)直接以氧气为氧化剂,邻苯二胺与苯甲醛反应成功合成了目标化合物2-苯基苯并咪唑。
(2)合成2-苯基苯并咪唑的最佳工艺条件为:n(苯甲醛)/n(邻苯二胺)=2,反应温度为45℃,反应时间为3h,甲醇/邻苯二胺的液固比(mL/g)为37.0(甲醇用量为40mL),产率最高可达71.2%。
[1] Mao ZZ,Wang ZY, et al.Research progress in the synthesis of benzimidazoles[J].Chinese Journal of Orginic Chemistry,2008,28(3):542-547.
[2] Li Y,Ma HQ,Wang YL.Progress in the synthesis and application of benzimidazoles and their derivatives[J].Chinese Journal of Orginic Chemistry, 2008, 28(2): 210-217.
[3] 张英,杨松,等.苯并咪唑类化合物杀菌活性的研究进展 [J].农药,2008,47(3):164-170.
[4] 叶佳,许孝良.苯并咪唑衍生物的合成研究进展[J].浙江化工,2012,43(10):21-24.
[5] 吴辉禄,高忆慈.超氧化物歧化酶模型化合物的合成、表征及其SOD活性研究[J].化学通报,2003(11):770-774.
[6] 张蓉,李锦堂,等.苯并咪唑类光电功能配合物的研究进展[J].材料导报,2011,25(7):61-64.
[7] 李莹莹,周永花,郭玉芳,等.苯并咪唑衍生物的合成改进[J].有机化学,2006,26(8):1097-1099.
[8] Lin Songnian, Yang Lihu.A simple and efficient procedure for the synthesis of benzimidazoles using air as the oxidant[J].Tetrahedron Letters, 2005(46):4315-4319.
[9] 于朝生,郑晓峰.自制氨基磺酸催化合成2-苯基苯并咪唑[J].化学工业与工程,2008,25(5):419-423.
[10] Ganesh, R, Jadhav, et, al.Ammonium metavanadate: A novel catalyst for synthesis of 2-substituted benzimidazole derivatives[J].Chinese Chemical Letters,2009(20): 292-295.
[11] Zhu Chenjie, Wei Yunyang.An Inorganic Iodine-Catalyzed Oxidative System for the Synthesis of Benzimidazoles Using Hydrogen Peroxide under Ambient Conditions[J].Chem.Sus.Chem., 2011, 4(8): 1082-1086.
[12] Singh M.P, Sasmal S, et al.Synthetic Utility of Catalytic Fe(III)/Fe(II) Redox Cycling Towards Fused Heterocycles:A Facile Access to Substituted Benzimidazole, Bi sbenzimidazoleandImidazopyridineDerivatives[J].Synthesis,2000(10):1380-1390.
[13] Kawashita Y, Nakamichi N, et al.Direct and Practical Synthesis of 2-Arylbenzoxazoles Promoted by Activated Carbon[J].Organic Letters, 2003(5):3713-3715.
[14] 于世树,李寅蔚.波谱分析法(第2版)[M].重庆:重庆大学出版社,1994:70-72.
[15] 詹东风,杜予民,钱保功.漆树酶催化氧化邻苯二胺及其产物结构鉴定[J].林产化学与工业,1991,11(1):13-16.
[16] 焦奎,孙刚,张书圣.OPD-H2O2-HRP伏安酶联免疫分析体系酶催化反应的研究[J].中国科学(B辑),1998,28(2):157-163.
