小尺寸铝纳米团簇的相变行为*
2013-09-27冯黛丽冯妍卉张欣欣
冯黛丽 冯妍卉 张欣欣
(北京科技大学热能与动力工程系,北京 100086)
(2012年8月22日收到;2012年12月23日收到修改稿)
1 引言
纳米团簇[1]通常由几个到几百个甚至上万个原子构成,直径在1—100 nm,这类零维材料表现出既异于单个原子,也不同于大块晶体的物理和化学性质.纳米团簇作为凝聚态物质的初始形态,在各种物质由原子分子向大块材料转变过程中起着至关重要的作用.而且,随着纳米材料的发展和相变蓄热/储能的需要,金属纳米团簇已成功应用于复合相变材料的制备,在优化、改善相变材料性能方面成效显著[2].因此,我们开始考虑将纳米团簇作为相变填充物,与纳米孔多孔基材进行组装,有望制备出高导热、高储热能力的纳米复合相变材料[3].
鉴于微观实验的高难度,目前金属纳米团簇相变行为的研究主要采用理论分析和分子动力学模拟(MD)的方法.如,Lewis等[4]开展的金团簇熔化和凝固的分子动力学模拟,显示出熔点和凝固点的差异,凝固过程发生了滞后.Reyes-Nava等[5]发现超过一定原子数的钠团簇在熔点附近出现了负热容现象.Kirchhoff等[6]采用紧束缚模型研究了金团簇相变的热膨胀系数等动力学特性.还有不少研究揭示了纳米团簇物性的尺度效应,如小颗粒金团簇的熔化[7]、铜团簇的熔点和凝固点[8],都具有团簇尺度依赖性.针对尺寸较大(直径大于5 nm)的金属团簇的相变,人们已尝试开展实验研究,如采用X射线衍射、扫描电镜、量热计、透射电镜等方法,测试了金[9]、银[10]、铟[11]、锡[12]和铅[13]等金属团簇的熔化特性.然而,现有的数值分析的结果还未能和实验值相互验证,彼此的结论也相去甚远.
综上所述,目前研究者更多是关注贵金属(如铂、金、银以及铜)的熔凝特性.而仅有的一些铝团簇相变的研究多采用实验手段[14,15],理论探讨有限,仅见Alavi等[16]的报道;且受实验条件的制约,铝团簇测试样品多为直径在几十纳米的“大”团簇.而纳米团簇若作为纳米复合相变材料的填充相,其尺度受限于基材纳米孔道的尺度、团簇在孔道内的填充率和填充状态,“小”纳米团簇将普遍存在.因此,本文针对小尺寸的铝纳米团簇,对其熔化、凝固特性开展分子动力学模拟与分析.团簇半径为几个纳米以下,原子数在13—555之间.本文工作以期为纳米复合相变材料中纳米相变填充物的热设计和制备提供一定理论和参数.

图1 Al N团簇初始结构和退火后结构
2 团簇结构的重构
分别构造出含不同原子数(即不同尺寸)的铝团簇,如图1所示.任意一个含N个原子的铝纳米团簇都是从具有面心立方体(FCC)结构的铝晶体中截取出来的.得到团簇的初始结构之后,为使其结构更加稳定、趋于真实结构,对其进行了退火处理.即对团簇升温到超过熔点几百K,再降回常温.退火后团簇结构更趋于有序化.
3 分子动力学模拟
3.1 势函数
本文采用原子嵌入模型(EAM)势函数[17],能有效描述金属如金、银、铜和铝等原子间的多体作用.作为一种半经验理论模型,其用于统计系统总能量的局域电子密度理论(LED)中的部分参数(如晶格常数、体变模量、结合能、弹性常数等)都是从实验中直接得到的.采用EAM势,含N个原子的体系总能量具有如下的表达形式:
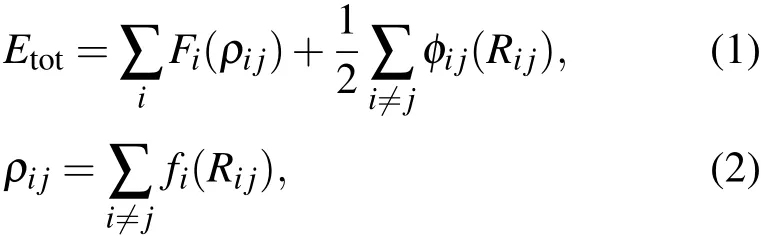
其中,Fi(ρij)为原子i嵌入时产生的嵌入能,ρij为除i外所有原子在i处产生的电子密度,φij(Rij)为原子i和 j间的作用势,Rij为i原子和 j原子间的距离,fj是原子 j在原子i处产生电子密度的函数.该模型建立在两个假设基础之上:1)原子i处的电子密度是由其余原子密度叠加求得;2)电子密度具有球对称性.
本文铝团簇具有FCC结构,则有
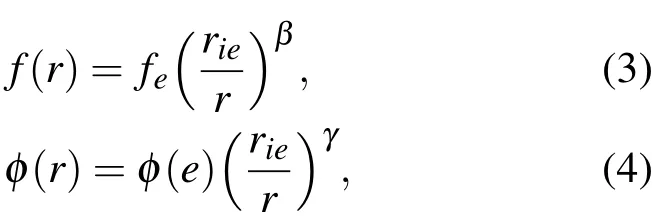
其中,

式中,r1e为平衡原子间距,β和γ为无量纲参数,EC为晶体结合能,Ω为原子体积,n为原子层数,Ni为第i层原子数,Ki为第i层原子半径与最近邻层的半径之比.所有的模拟参数都取自参考文献[17].
3.2 熔、凝点计算方法
本文将采用势能温度函数法[18],结合热容-温度曲线,来确定团簇的熔、凝点,其中热容的计算通常有两种统计学方法.第一种方法是由内能的变化来计算热容:

其中,kB为玻尔兹曼常数,NVT表示正则系综,代表了许多具有相同温度的体系的集合,系综内每个体系的粒子数和体积都是相同的.第二种方法则是通过内能对温度的导数求得热容

本文采用第二种方法来获得热容.
需要指出的是,除了势能温度函数法,纳米团簇熔、凝点的确定方法还包括径向分布函数法[18]和键对分析技术[18]等.其中,径向分布函数法通过描述一个原子周围其他原子按距离的分布情况能够得到团簇熔点的大概范围,直观看出体系的变化趋势;键对分析法则能够给出团簇的微观构型随温度的变化,也能较好地找出团簇的熔点.但相较而言,势能温度函数法还是最常用也是更为精确的一种方法.
3.3 模拟计算过程
在无周期性边界条件下的NVT下,开展铝团簇势能的分子动力学模拟.运动方程的求解采用Verlet蛙跳法(leap-frog),时间步长取2 fs.团簇完成退火之后得到的基态结构,其初始速度置为零.先设定一个初始温度,在该温度下每一个原子的初速度按Maxwell分布随机给定,进而实现团簇温度的改变.每次升温(降温)温度间隔为100 K,到熔点或者凝固点附近降为10—1 K.为保证体系在每个温度点上达到热力学平衡,每个温度点至少进行2×106次计算.而熔点和凝固点附近因其能量不稳定,所需迭代步数比其他位置要长很多(至少2×107次).
4 结果与讨论
本文模拟得到了原子数不同的9组铝纳米团簇的熔、凝点,如表1所示.其中N与r有如下关系:r∝ N1/3.
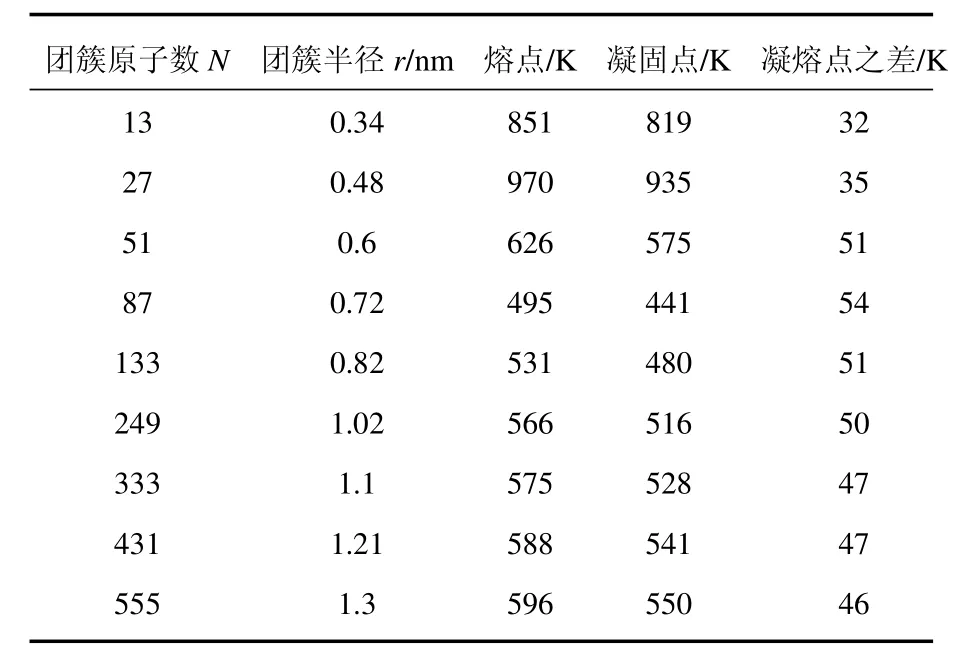
表1 不同原子数的铝纳米团簇的熔点和凝固点
以原子数N=249的铝团簇Al249为代表,图2和图3分别给出了团簇在热化和冷凝过程中势能函数、热容随温度变化的曲线.从图2可见,在热化和冷凝过程中,势能均出现了明显的跳跃,跳跃所对应的温度即为相变温度;而在这个跳跃前后,势能随温度变化基本为线性变化.由势能温度曲线来确定熔、凝点的方法比较直观,但精确度不是太高,跳跃覆盖了一个温度范围,无法锁定某一个温度值.因此,需要进一步分析热容的变化来确定准确的熔、凝点,如图3所示.通过该热容-温度曲线,准确锁定Al249团簇的熔点和凝点分别为571和516 K.

图2 Al249纳米团簇的势能-温度曲线

图3 Al249纳米团簇的热容-温度曲线
按照上述方法得到不同原子数的团簇的熔凝点,如表1和图4所示.图4展示了铝团簇熔凝点随团簇尺寸的变化.
当小团簇尺寸相对较大时(团簇原子数N>80),将本文的模拟结果与文献[16]的模拟结果进行了比较.从图4可见,两者相近;考虑到本文与文献[16]分子动力学模拟所选用的势函数、步长、统计方法等的不同,可认为两种模拟结果互为验证.此外可见,团簇的熔凝点均低于铝块体的熔凝点(933 K);且随着原子数的减少,纳米团簇的熔凝点也相应降低.这个结果充分体现了纳米团簇熔凝点的尺寸效应,即熔凝点随纳米晶体尺寸的减小而降低,与前人所做的研究工作结论相一致.产生这种现象的原因是由于表面原子具有的表面能与团簇的总能量比例的变化.球形颗粒比表面积(表面积/体积)与直径成反比,随着颗粒直径的减小比表面积将会显著增加,表面原子在整个团簇的总原子数中占据的比例越大,表面能的比重也迅速增大.表面原子的受力环境和结合能与内部原子不同,表面原子周围缺少足够多的相邻原子,导致原子配位不足.因此表面原子极易与其他原子结合,具有很大的化学活性.晶体尺度纳米化的过程伴有这种活性表面原子的增多,导致表面能大大增加.故而团簇的活性增大,结构不稳定,打破这种结构所需要的能量也就越低,也就是说,纳米团簇熔化所需要增加的内能比块体材料熔化所需的能量小得多,即表现为纳米团簇熔点相对块材熔点的急剧降低,且具有尺度依赖性.
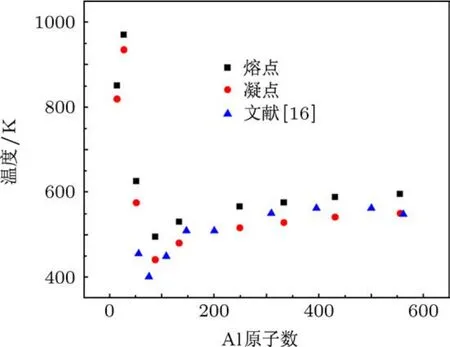
图4 小尺度铝纳米团簇熔凝点随原子数N的变化
而当团簇具有更小尺寸时(如本文的N=13,27,51),相应的熔凝点则显著高于本文模拟范围内的大团簇,且不同于大团簇,熔凝点的尺寸依赖性呈现出非单调的变化.分析认为,这是因为极小尺寸团簇的束缚能不随尺寸单调变化.这与两个壳层效应密切相关:一个是几何壳层效应,小尺寸团簇几个原子的改变即可能导致团簇结构的巨变,有可能形成具有非常稳定结构的规则多面体,如十四面体、二十面体,而打破这种极其稳定结构所需要的能量很高,即反映为熔点的显著升高;另一个是与之相似的电子壳层效应,电子在能量空间的排布决定团簇的稳定性,满壳层排布时,团簇最为稳定.这些效应决定着团簇稳定性的最大值和最小值,这也就决定了团簇的熔点不和尺寸大小成单调变化,尤其当N=27时,熔点达到970 K,高出铝块材熔点近40 K,可归因于其几何体的高度重构,导致结构异常稳定,进而表现出极高的热力学稳定性,从而具有高熔点.极小尺寸团簇熔点的这种非单调变化的尺度依赖性以及结构重构导致熔点高于块材的现象,在前人对小团簇金属锡、铅、铂、镍等的研究中有过类似报道[19,20],10—30个原子的锡纳米团簇中,部分团簇熔点比块材高出约50 K.包含12—34个原子的铅、铂及镍金属团簇熔点与尺寸关系则呈现无规律的异常变化,同时也出现高于晶体熔点的现象.这与本文的研究结果是相一致的.目前,关于小尺寸团簇熔点异常的现象还未能有明晰的物理解释,究竟是几何构型还是电子壳层排布对团簇的稳定性影响更大,还未有定论.究竟何种机制是导致熔点异常的关键,以及是否存在某个原子数N,为熔点由随尺寸无规律变化到单调变化的一个转折点,将是后续研究的重点.
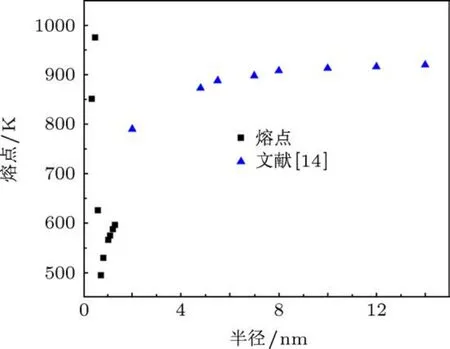
图5 大、小尺度范围内的铝纳米团簇熔点随半径的变化
根据经典的热力学模型,可根据Hanszen公式[21]从理论上计算金属纳米团簇熔点随其尺寸的变化规律,但该热力学构架关系仅适用于“大”的纳米团簇,部分参数需由实验获得.Lai等[14]通过实验得出了“大”尺寸铝团簇的熔点随半径变化的规律,如图5所示.可见,大尺寸团簇的熔点与半径呈单调变化,随尺寸增加,熔点由较快的增长逐渐变得缓慢,最终趋近于铝块材的熔点.而本文所研究的“小”尺寸团簇在无法实验测量以及Hanszen理论公式不适用的前提下,采用模拟方法获得的熔点参数,是对纳米团簇“小”尺寸领域的合理补充,丰富了铝团簇的相变规律.
本文模拟结果还显示,对于同一个团簇,其熔点和凝固点并不一致.在本文研究的原子数范围内,团簇的凝固点均低于其熔化温度,见表1和各图.也就是说,纳米尺寸的团簇由液相到固相的转变滞后于固相到液相的转变.凝固滞后现象已从理论和实验中得以证实[4,22],这也证明了物质从有序(固体)到无序(液体)比无序到有序更容易.这在宏观相变机理中也是成立的,表现为金属凝固过程中的过冷现象.对于金属块材,过冷度(熔化温度与实际凝固温度之差)是金属液体结晶的必要条件.为了得到过冷度,系统自由能必须减少,也就是内能必须减少.同时,在液体中形成固体时,会产生表面能.于是,在整个凝固形核的过程中,自由能在减小,表面能在逐渐增大,只有在自由能减小的趋势大于表面能增大的趋势时,形成的晶核才能稳定,凝固才能持续进行.而金属在纳米团簇状态下,表面能急剧增大,因此要达到成核条件就比块材状态下困难了很多,凝固点与熔点之差也就显著增加.铝块体凝固时的过冷度仅为几K,而本文研究的铝纳米团簇实际凝固温度与熔化温度之差在几十K.从这个意义上来分析,团簇的熔点比块材的熔点更好地反映了热力学的固-液相变点的本质.
5 结论
本文选用原子嵌入势能模型,对小尺寸铝纳米团簇(包含原子数13—555)的熔化、凝固过程进行分子动力学模拟,基于势能-温度与热容-温度曲线预测热力学性质,并利用表面效应、小尺寸效应等纳米材料领域的基础理论开展现象分析.
研究结果表明:不同于大尺寸纳米团簇,在本文模拟的小尺寸范围内,当团簇原子数大于80,铝纳米团簇的熔点和凝固点均随团簇尺度减小而降低,且远低于块体材料的相变点;当原子数小于80,熔、凝点随尺寸呈无规律变化;当原子数为27时,团簇熔点高于块材熔点近40 K.此外,纳米团簇具有明显的凝固滞后现象.本文工作是对纳米团簇小尺寸领域的补充,丰富了铝团簇的相变规律,以期为复合相变材料中纳米相变填充物的热设计和制备提供一定理论和参数.
[1]Cao M S,Guan C B,Xu J Q 2001 Introduction of Nano Materrals(Haerbin:Harbin Institute of Technology Press)(in Chinese)[曹茂盛,关长斌,徐甲强2001纳米材料导论(哈尔滨:哈尔滨工业大学出版社)]
[2]Song QW,Li Y,Xing JW,Hu JY,Marcus Y 2007 Polymer 48 3317
[3]Huang CL,Feng Y H,Zhang X X,Li J,Cui L,Wang G,Yang M 2011 Int.J.Thermophys.DOI:10.1007/s10765-011-1076-x
[4]Lewis L J,Jensen P,Barrat JL 1997 Phys.Rev.B 56 2248
[5]Reyes-Nava JA,Ignacio L G,Michaelian K 2003 Phys.Rev.B 67 165401
[6]Kirchhoff F,Mehl M J,Papanicolaou N I,Papaconstantopoulos D A,Khan FS 2001 Phys.Rev.B 63 195101
[7]Ercolessi F,Andreoni W,Tosatti E 1991 Phys.Rev.Lett.66 911
[8]Wu Z M,Wang X Q,Yang Y Y 2007 Chin.Phys.16 405
[9]Buffat P,Borel JP1976 Phys.Rev.A 13 2287
[10]Castro T,Reifenberger RE 1990 Phys.Rev.B 42 8548
[11]Coombes CJ1972 J.Phys.Metal Phys.2 441
[12]Wronski CRM 1967 Br.J.Appl.Phys.18 1731
[13]Peters K F,Cohen JB,Chung Y 1998 Phys.Rev.B 57 13430
[14]Lai SL,Carlsson JRA,Allen L H 1998 Appl.Phys.Lett.72 1098
[15]Sun J,Simon SL 2007 Thermochim.Acta 463 32
[16]Alavi S,Thompson D L 2006 J.Phys.Chem.A 110 1518
[17]Johnson RA 1989 Phys.Rev.B39 12554
[18]Zhao L J,Li B,Gao H,Li D S 2009 J.Chongqing Univ.32 67(in Chinese)[赵良举,李斌,高虹,李德胜2009重庆大学学报32 67]
[19]Lee Y J,Lee EK,Kim S 2001 Phys.Rev.Lett.86 999
[20]Luo Q,Wang X Q,He H D,Wu ZM,Xiao X Y 2006 Journal of Synthetic Crystals 35 351(in Chinese)[罗强,王新强,何焕典,毋志民,肖绪洋2006人工晶体学报35 351]
[21]Hanszen K J1960 Z.Phys.157 523
[22]Kofman R,Cheyssac P,Aouaj A,Lereah Y,Deutschera G,Ben-Davida T,Penissonc JM,Bourret A 1994 Surf.Sci.303 231
