大气颗粒物高时间分辨采样和定量测量技术
2013-09-22霍俊涛许涵乾
吴 秀,霍俊涛,许涵乾,陈 宏,杨 新
(复旦大学 环境科学与工程系,上海200433)
大气气溶胶是空气与悬浮在大气中的颗粒组成的多相体系,通常是指空气动力学直径为0.003~100μm的固体或(和)液体微粒[1,2].近年来,气溶胶颗粒会影响到人体健康、空气质量、能见度以及气候变化而受到人们的广泛关注[2].气溶胶的理化性质和其环境效应及健康效应取决于它们的来源以及在大气中的传输和转化过程.因此,要想理解气溶胶的环境和气候效应,就必须对其化学组分进行高时间分辨的分析测量.
以细颗粒气溶胶PM2.5中含量最丰富的二元羧酸-草酸为例.草酸在大气中的浓度介于几到数百ng/m3,而城市中的草酸含量更高,一般都在几百个ng/m3以上[3,4].研究PM2.5中的草酸浓度及其变化规律有助于了解二次气溶胶的形成机制和变化规律,对于进一步研究大气颗粒物的物理化学特性有着十分重要的意义.草酸的来源可以分为一次源和二次源,重要的一次源包括生物质燃烧[5,6],汽油和重油的燃烧[7,8].其他如食品烹饪[9,10],香烟[11]等都构成了草酸的来源.另一方面,二次生成的草酸机理包括挥发性有机物(VOC)气相光化学反应及由于人为源和生物排放源进一步引起的气相和粒相的转化[12,13],气溶胶表面的非均相反应[14-16],及云内过程[17-20]等.气相有机物和某些氧化物种如苯系物和长链的有机酸的二次化学反应也会产生草酸[21,22].由于气象条件和环境条件的复杂性,生成草酸的主要机理也因时因地而异.正因为草酸具有多种不同的来源,在大气中的多个环节中又会发生以上所说的各种复杂变化,因而要想准确描述大气中的草酸的变化规律,也相对困难.
鉴于气溶胶中草酸的重要性及复杂性,多个课题组对气溶胶中的草酸进行了不同层面深入的研究[23-26],但这些检测方法均不是高时间分辨的方法,对突发事件,从技术手段上要实现对草酸的高时间分辨的定量分析是比较困难的.目前已有一些研究小组试图对草酸做一些相对高时间分辨的研究[27-29].Miyazaki[27]在印度做了草酸等昼夜变化的研究,发现日间水溶性气溶胶总量上升,而夜间草酸含量上升,白天主要是发生了光化学反应过程,而晚上是由于云过程及生物排放的综合结果.Kawamura小组[28]报道了3h分辨的多种有机酸的研究,8:00~11:00时间内下降到35%,而17:00~20:00时间内又增加到58%,结果说明1d之内气溶胶中草酸变化也是非常复杂的.
要想获得高时间分辨的化学信息,首先要实现高时间分辨的采样.时间分辨的颗粒物采样器已被运用了很多年,但一般都是采集总颗粒物(TSP).针对细颗粒物的时间分辨微量采样未见报道.由于细颗粒物相对质量浓度偏低,一般的分析技术都需要大容量长时间不间断采样才能够对样品进行化学组分分析.这样的分析结果是一天甚至几天的平均结果,无法追踪到细颗粒气溶胶成分随时间的变化.草酸的检测主要有两种方法:离子色谱(IC)和气相色谱质谱联用(GC-MS).还有其他一些方法如结合红外光谱,毛细管电泳等[30-32].这些方法,均对样品总量有较高要求,一般都要进行预处理才能完成后续步骤的操作,费时费力.无需样品前处理的,简单快速同时具有很高灵敏度的检测方法是气溶胶组分分析的发展趋势.
本课题组首次将电喷雾解析电离质谱(DESI-MS)技术应用于气溶胶化学组分分析[33,34].该技术可对微克级样品进行定量分析,且无需样品前处理,使气溶胶微量样品分析成为可能.本文报道了我们自行研制的高时间分辨微量采样技术,并将细颗粒物微量采样器与快速高灵敏的检测手段DESI-MS技术相结合,实现对大气环境中气溶胶化学组分高时间分辨的定量分析,有助于对相关大气化学过程进行更为深入的研究.
1 实验部分
1.1 实验装置
(1)PM2.5高时间分辨采样器 自行研制的高时间分辨气溶胶采样仪是由单板机控制的便携式小型仪器,总重量不超过10kg,便于携带.仪器包括了空气动力学部分、计算机控制部分、采样器部分和真空部分.其中的核心部分——由计算机控制的微量采样器如图1(a)所示.采样的基本原理是由真空泵带动的滤膜采样.滤膜为盘在传动装置上的宽度2.5cm的石英纤维纸带.抽滤装置与纸带的接触时间(即采样时间)和滤膜传动装置的转动速度均由单板机控制.采样器现配有1个可以拆卸的 PM2.5切割头,可实现 PM2.5和TSP采样转换.采样泵的抽速为16.7L/min.滤膜采样面积为直径1cm2的圆斑(见图1(b)).这样大流量、小面积的采样可以在较短时间内在单位面积上富集足够的样品,采样时间可以大大缩短,这是实现高时间分辨测量的基础.DESI-MS样品分析面积仅需几平方毫米,对样品总量要求不大.可以根据实际大气污染的状况以及不同化学物质的含量调整采样时间,以获得达检测限的样品量.仪器设定最小采样时间为15min,最大为6h.采样器中有可调温控装置,可以减少水分在测量结果中的干扰.另外,此采样仪中集成了温湿度传感器,可以在采样的同时记录下温湿度及大气压力的变化,为后续的数据处理及分析提供在线的数据支持.
(2)DESI-MS测量 DESI-MS的原理在文献[35,36]中已有报道.采用此电离源时,电喷雾产生的带电液滴及离子在常压下直接冲击被分析物表面,吸附在表面的待测物受到带电离子的撞击,在带电雾滴及鞘气的共同作用下,以离子的形式解析出来,然后通过质谱仪的采样口进入质量分析器,获得的质谱图与常规电喷雾质谱图十分相似,可以得到带有单个或多个电荷的分子离子.
本实验是在Thermo Finnge LCQ-Advantage质谱仪上进行.DESI离子源由美国Prosolia公司生产,主要由4部分组成:雾化器三维微调部件、表面三维微调部件、摄相和光源部件,固定及支撑部件.离子源通过控制器与质谱仪和计算机相连,具有准确控制样品几何位置和连续自动进样的功能.其中,几何参数是影响样品离子化效率的重要参数,准确定位可明显提高实验结果的重现性;连续自动进样,则实现了高通量分析.

图1 单板机控制的微量采样器(a)和4h采样的斑点(b)Fig.1 Trace amount aerosol sampler controlled by single-board computer(a)and sampled spots after four hours sampling(b)
1.2 样品的采集
大气气溶胶高时间分辨微量采样器位于复旦大学环境科学系楼楼顶,四周为复旦大学校园教学区,500m范围内有一条交通主干道.采样时间为2011年1月6~7日,3月18~19日和4月18~19日.采样器进行昼夜连续采样.1月6~7日采样时间为:白天12:00至次日10:00.3月18~19日采样时间为下午13:00至次日9:00,4月18~19日采样时间为上午12:00至次日10:00.其中,1月6~7日和4月18~19日的采样频率为每2h采一个样;3月18~19日每小时采1个样.采样器使用PM2.5切割头.
1.3 实验操作与仪器参数
DESI-MS所测质量范围:50~500;总气体流量:0.8MPa;喷雾电压:5.8kV;喷雾溶剂流速:2.5~12.5μL/min;质谱入口与样品表面的距离:1~2mm;喷雾针尖与样品表面的距离:2~3mm;喷雾针与样品表面的角度:55°;样品表面与质谱入口毛细管的角度:35°;毛细管电压:-10V;毛细管温度:110℃;透镜电压:-50V.
用分析天平准确称取一定量的H2C2O4·2H2O固体置于5mL琥珀色安培瓶中,用甲醇溶解配成草酸浓度为104μg/mL的标准储备液,然后依次用甲醇稀释成一系列浓度的标准工作溶液,4℃冰箱保存备用.分别用移液枪量取不同浓度的10μL标准工作溶液,滴加到面积为1cm2的滤膜上,进行定标实验.质谱质量范围设置在50~600(m/z),连续扫描1min,通过Xcalibur软件界面控制以获得数据(0.6s/spectrum).当数据稳定后,每个样品测6次,每次持续时间为1min.优化后的实验参数能增加测试时的草酸信号强度.喷射溶液是影响目标离子信号强度的关键因素.用50/50(V/V)甲醇/水溶液和含1%氨水的甲醇溶液,在负离子检测模式下能获得较强的草酸信号,而相比之下,后者的信号更强.因此选用含1%氨水的甲醇溶液作为喷雾溶剂.
2 结果与讨论
2.1 PM2.5浓度水平
滤膜上细颗粒物(PM2.5)的质量通过差重法测得.采用灵敏度为1μg的电子微量天平进行测量.采样前滤膜在干燥器内放置24h,用天平称量单位长度重量,放回干燥器1h后再称重.将已恒重好的滤膜用于采样.采样结束后,取出采样滤膜,将有样品部分均匀剪成相同长度,尘面2次对折,放入铝箔中并做好采样记录.同采样方法称量滤膜质量.按下式计算颗粒物的质量浓度:

其中,C为 PM2.5质量浓度(μg/m3);G1为空白滤膜的质量(g);G2为采样后滤膜的质量(g);Yr为采样时间与流量之积,并换算成标准状态下的采样体积(m3).图2显示的是2011年1月6~7日,采样器所测的2h分辨的PM2.5的质量浓度,并与上海市环保局所公布的PM10数据进行对比.数据具有很好的相关性.
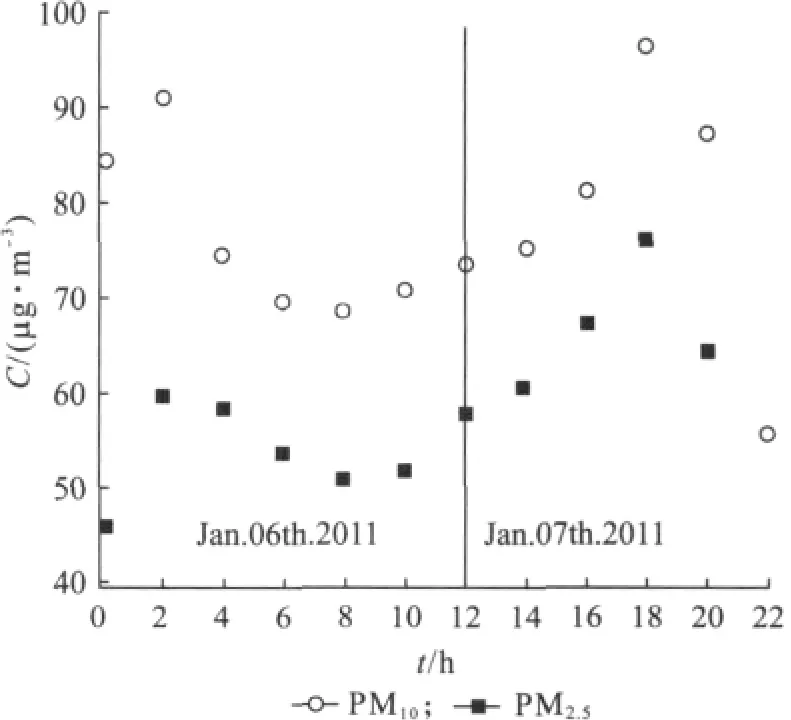
图2 2011年1月6~7日微量采样器所测PM2.5质量浓度与上海市环保局发布的PM10数值对比Fig.2 Comparison of PM2.5mass concentration measured by the sampler and the PM10mass concentration released by the Shanghai Environmental Protection Bureau on January 6th-7th,2011
2.2 大气气溶胶中有机酸的定性分析
图3是本实验所采集的大气气溶胶样品的2张DESI-MS图谱.
从图3中可以看出,采用1%氨水甲醇溶液作为喷雾溶剂,在优化条件下,气溶胶中的有机酸被选择性检测出来.利用DESI-MS分析气溶胶中草酸,还检测到了丙二酸,丁二酸,苯甲酸,甲基苯甲酸,正己酸,正癸酸,硬脂酸等有机酸的信号.说明DESI-MS可用于多种有机酸的同时分析,检测范围较广.
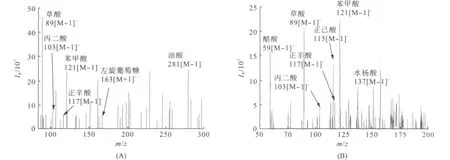
图3 负离子模式下大气气溶胶中有机酸的DESI图谱Fig.3 The DESI spectrum of organic acids in atmospheric aerosol in negative ion mode
2.3 草酸质量浓度的定量测量
为保证数据的准确性和精确度,每次测样品之前都要做相应的标准曲线.一般配置7个不同浓度(1pg~1μg)的草酸标准溶液并在测定其信号强度后画出标准曲线,草酸浓度的坐标轴用对数刻度处理.标准溶液配置完后直接滴在滤膜上测量,每个浓度测6次,每次1 min.如果目标物质的信号强度达到至少是背景值的3倍,那么其信号就可看作高于检出限.图4为典型的标准曲线,草酸浓度与其信号强度之间呈现出良好的线性关系(R2>0.99),相对标准偏差小于10%.检测限约为1pg/mm2.
将采集有气溶胶样品的滤膜直接放入DESI-MS检测草酸信号,无需任何预处理.草酸的浓度可根据标准曲线经换算获得.换算公式如下(以图4所示标准曲线为例):
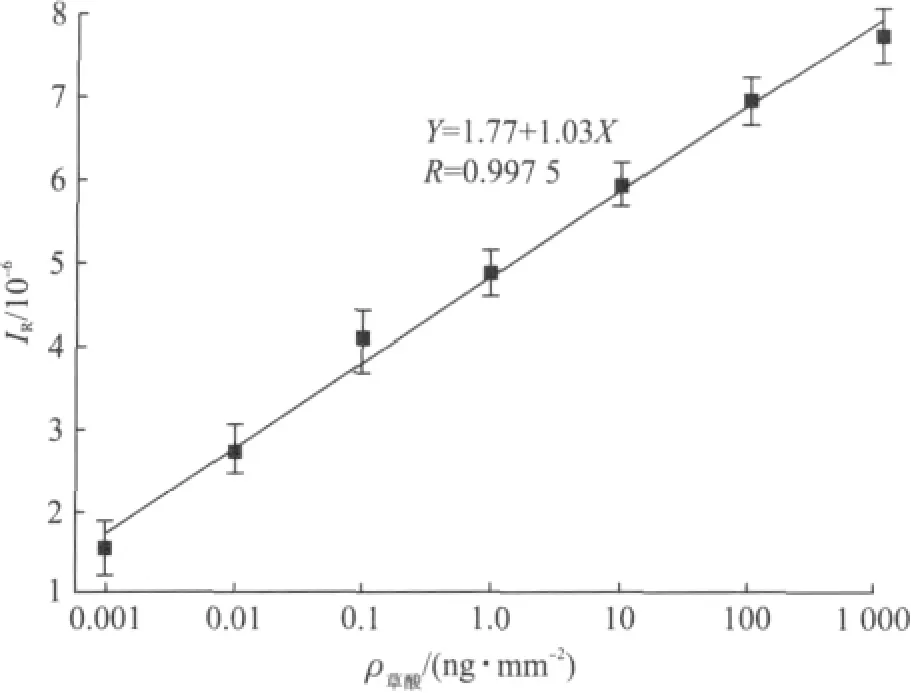
图4 DESI-MS测量草酸标准曲线Fig.4 The calibration curve for DESI-MS oxalic acid measurements

其中,C为草酸的质量浓度(ng/m3);y为信号强度;S为样品的面积,本实验中S为113.0mm2;V为每个采集样品对应的抽取空气体积,当采样时间为1h时,V为1m3.代入式(2),得到每个草酸样品的质量浓度,作出草酸质量浓度日变化曲线.
2.4 草酸质量浓度的日变化分析
3个采样时段的小时或2h分辨的PM2.5中草酸质量浓度如图5所示.1月6~7日,天气多云,风力4~5级,温度低,湿度大,草酸的质量浓度总体趋势是白天高于夜晚.峰值出现在7日早上8:00,最低值出现在6日晚上12点.1月6日中午,草酸的浓度变化趋势与温度变化成正相关,此段时间内的高值可能是由于光氧化反应强烈的结果.2005年,Kawamura分析了二元羧酸形成与环境温度、太阳辐射以及氧化物浓度的关系,指出由于气温上升,光化学反应增强促使一些草酸的前驱物分解形成草酸[28].下午14:30后至16:30时,草酸质量浓度开始下跌,可能是因为环境温度降低、光照减弱,从而抑制了草酸的形成.然而16:00之后,草酸浓度又开始缓慢上升.Kawamura也发现了同样的现象,他认为下午草酸浓度的上升是大气中的氧化物(如硝基)氧化物草酸前驱物生成草酸所致[28].1月7日4:00起,草酸浓度急剧上升,至8:00达最大值.这一峰值的形成一方面是PM2.5质量浓度上升所致(见图2),另一方面可能与早晨边界层较低和出行高峰期机动车尾气排放的结果叠加有关.

图5 PM2.5中草酸质量浓度的h或2h分辨测量结果Fig.5 The measured oxalic acid mass concentration with one or two hours time resolution in PM2.5
3月18~19日的草酸浓度变化与1月6~7日的结果非常相似,均呈白天高,晚上低.而4月18~19日的结果则不同.实际上,4月18日2:00的高值(约400)和20:00左右的晚高峰,与前两个采样时段的结果颇为相似.但从19日凌晨起,草酸浓度突然上升至1 200ng/m3左右,随后缓慢下降.草酸在夜间出现高值的原因可能有:1)夜间空气湿度较大,有利于液相反应发生;2)夜间大气扩散能力弱,污染物难以扩散稀释.但这些都难以解释出现在凌晨的高峰,不排除有污染传输所致.从这三个时段的分析不难看出,PM2.5中草酸由于来源众多,盲目的日平均和月平均浓度统计会因大量丢失大气化学信息而失去科学意义.
[1]唐孝炎,张远航,邵 敏.大气环境化学[M].北京:高等教育出版社,2006.
[2]Li S M,Winchester J W.Geochemistry of organic and inorganic-ions of late winter arctic aerosols[J].AtmosphericEnvironment,1989,23(11):2401-2415.
[3]Rohrl A,Lammel G.Low molecular weight dicarboxylic acids and glyoxylic acid:Seasonal and air mass characteristics[J].EnvironmentalScience&Technology,2001,35(1):95-101.
[4]Yang H,Yu J Z,Ho S S H,etal.The chemical composition of inorganic and carbonaceous materials in pm2.5in nanjing,china[J].AtmosphericEnvironment,2005,39(20):3735-3749.
[5]Fang M,Zheng M,Wang F,etal.The solvent-extractable organic compounds in the indonesia biomass burning aerosols-characterization studies[J].AtmosphericEnvironment,1999,33(5):783-795.
[6]Simoneit B R T.Biomass burning a review of organic tracers for smoke from incomplete combustion[J].AppliedGeochemistry,2002,17(3):129-162.
[7]Rogge W F,Hildemann L M,Mazurek M A,etal.Sources of fine organic aerosol.2.Noncatalyst and catalyst-equipped automobiles and heavy-duty diesel trucks[J].EnvironmentalScience&Technology,1993,27(4):636-651.
[8]Schauer J J,Kleeman M J,Cass G R,etal.Measurement of emissions from air pollution sources.2.C-1 through C-30organic compounds from medium duty diesel trucks[J].EnvironmentalScience&Technology,1999,33(10):1578-1587.
[9]Rogge W F,Hildemann L M,Mazurek M A,etal.Sources of fine organic aerosol.1.Charbroilers and meat cooking operations[J].EnvironmentalScience&Technology,1991,25(6):1112-1125.
[10]Schauer J J,Kleeman M J,Cass G R,etal.Measurement of emissions from air pollution sources.1.C-1through C-29organic compounds from meat charbroiling[J].EnvironmentalScience&Technology,1999,33(10):1566-1577.
[11]Rogge W F,Hildemann L M,Mazurek M A,etal.Sources of fine organic aerosol.6.Cigarette-smoke in the urban atmosphere[J].EnvironmentalScience&Technology,1994,28(7):1375-1388.
[12]Kerminen V M,Ojanen C,Pakkanen T,etal.Low-molecular-weight dicarboxylic acids in an urban and rural atmosphere[J].JournalofAerosolScience,2000,31(3):349-362.
[13]Kawamura K,Ikushima K.Seasonal-changes in the distribution of dicarboxylic-acids in the urban atmosphere[J].EnvironmentalScience&Technology,1993,27(10):2227-2235.
[14]Kerminen V M,Teinila K,Hillamo R,etal.Size-segregated chemistry of particulate dicarboxylic acids in the arctic atmosphere[J].AtmosphericEnvironment,1999,33(13):2089-2100.
[15]Mochida M,Kawamura K,Umemoto N,etal.Spatial distributions of oxygenated organic compounds(dicarboxylic acids,fatty acids,and levoglucosan)in marine aerosols over the western pacific and off the coast of east asia:Continental outflow of organic aerosols during the ace-asia campaign[J].Journalof GeophysicalResearch,108(D23):243-248.
[16]Turekian V C,Macko S A,Keene W C.Concentrations,isotopic compositions,and sources of sizeresolved,particulate organic carbon and oxalate in near-surface marine air at bermuda during spring[J].JournalofGeophysicalResearch-Atmospheres,108(D5):4157-4162.
[17]Faust B C.Photochemistry of clouds,fogs,and aerosols[J].EnvironmentalScience&Technology,1994,28(5):A217-A222.
[18]Blando J D,Turpin B J.Secondary organic aerosol formation in cloud and fog droplets:A literature evaluation of plausibility[J].AtmosphericEnvironment,2000,34(10):1623-1632.
[19]Yao X H,Fang M,Chan C K.Size distributions and formation of dicarboxylic acids in atmospheric particles[J].AtmosphericEnvironment,2002,36(13):2099-2107.
[20]Yao X H,Lau A P S,Fang M,etal.Size distributions and formation of ionic species in atmospheric particulate pollutants in beijing,china:2-dicarboxylic acids[J].AtmosphericEnvironment,2003,37(21):3001-3007.
[21]Seinfeld J H,Pandis S N.Atmospheric chemistry and physics:From air pollution to climate change[M].New York:Wiley-Inter Science,1998.
[22]Schauer J J,Fraser M P,Cass G R,etal.Source reconciliation of atmospheric gas-phase and particlephase pollutants during a severe photochemical smog episode[J].EnvironmentalScience&Technology,2002,36(17):3806-3814.
[23]Miyazaki Y,Kawamura K,Sawano M.Size distributions and chemical characterization of water-soluble organic aerosols over the western north pacific in summer[J].JournalofGeophysicalResearch-Atmospheres,2010,30(25):115-119.
[24]Ho K F,Cao J J,Lee S C,etal.Dicarboxylic acids,ketocarboxylic acids,and dicarbonyls in the urban atmosphere of china[J].JournalofGeophysicalResearch-Atmospheres,2007,28(35)112-116.
[25]Wang G H,Niu S L,Liu C,etal.Identification of dicarboxylic acids and aldehyde of PM10and PM2.5aerosols in nanjing,china[J].AtmosphericEnvironment,2002,36(12):1941-1950.
[26]Ho K F,Lee S C,Cao J J,etal.Dicarboxylic acids,ketocarboxylic acids and dicarbonyls in the urban roadside area of hong kong[J].AtmosphericEnvironment,2006,40(17):3030-3040.
[27]Miyazaki Y,Aggarwal S G,Singh K,etal.Dicarboxylic acids and water-soluble organic carbon in aerosols in new delhi,india,in winter:Characteristics and formation processes[J].Journalof GeophysicalResearch-Atmospheres,2009,36(15)114-119.
[28]Kawamura K,Yasui O.Diurnal changes in the distribution of dicarboxylic acids,ketocarboxylic acids and dicarbonyls in the urban tokyo atmosphere[J].AtmosphericEnvironment,2005,39(10):1945-1960.
[29]牛红云,陈 洁,王格慧,等.南京市大气气溶胶中二元羧酸昼夜变化研究[J].环境科学研究,2005,18(6):23-26.
[30]Norton R B,Roberts J M,Huebert B J.Tropospheric oxalate[J].GeophysicalResearchLetters,1983,10(7):517-520.
[31]Kawamura K,Kaplan I R.Motor exhaust emissions as a primary source for dicarboxylic-acids in losangeles ambient air[J].EnvironmentalScience&Technology,1987,21(1):105-110.
[32]Obrien R J,Holmes J R,Bockian A H.Formation of photochemical aerosol from hydrocarbons-chemical reactivity and products[J].EnvironmentalScience&Technology,1975,9(6):568-576.
[33]Chen H,Li M,Zhang Y P,etal.Rapid analysis of svoc in aerosols by desorption electrospray ionization mass spectrometry[J].JournaloftheAmericanSocietyforMassSpectrometry,2008,19(3):450-454.
[34]Li M,Chen H,Yang X,etal.Direct quantification of organic acids in aerosols by desorption electrospray ionization mass spectrometry[J].AtmosphericEnvironment,2009,43(17):2717-2720.
[35]Takats Z,Wiseman J M,Gologan B,etal.Mass spectrometry sampling under ambient conditions with desorption electrospray ionization[J].Science,2004,306(5695):471-473.
[36]Cooks R G,Ouyang Z,Takats Z,etal.Ambient mass spectrometry[J].Science,2006,311(5767):1566-1570.
