大肠杆菌工程菌ptsG基因敲除及其缺陷株混合糖同型乙醇发酵
2013-09-04严涛赵锦芳高文慧王金华王永泽赵筱周胜德
严涛,赵锦芳,高文慧,王金华,王永泽,赵筱,周胜德
工业发酵湖北省协同创新中心 湖北工业大学生物工程学院,湖北 武汉 430068
在底物为六碳糖和五碳糖的情况下,大肠杆菌会优先利用速效碳源 (即六碳糖),然后再利用迟效碳源 (即五碳糖),这种现象称为碳代谢产物抑制 (CCR)[1],这是由于相关酶的活动和基因表达的情况下,抑制了细胞运输其他碳源的能力[2-4]。所有的这些现象与磷酸转移酶系统 (PTS)密切相关[5],此系统对于菌体具有以下生理学意义:碳水化合物转运和代谢、分解物代谢阻遏、碳源储备、协调碳氮代谢平衡等[6]。PTS系统包括EI、HPr以及EIIs,前两者为胞质可溶型蛋白,分别由pts H和ptsⅠ编码,后者多为蛋白复合体,对碳水化合物具有特异性,包括EIIMan、EIIFru、EIIBgl、EIIAGlc、EIICBGlc等[6],其中crr基因编码的EIIAGlc以及ptsG基因编码的EIICBGlc在葡萄糖磷酸化转运中起主要作用[7],其中由ptsG基因编码的酶EIICBGlc在葡萄糖的跨膜转动中具有重要作用[8]。
关于ptsG基因缺陷菌株与大肠杆菌Escherichia coli发酵的研究在国内也有相关报道[9],Li等[1]构建了大肠杆菌PTS缺陷株,即敲除ptsG基因,使大肠杆菌同时能利用混合糖发酵来生产聚羟基脂肪酸酯 (PHAs);韩聪等[10]以Escherichia coliDH5α、JM109为出发菌株,在重组酶的作用下,构建ptsG基因缺陷株,结果发现,缺陷菌株的最高菌密度远高于对照菌株。重组蛋白肿瘤坏死因子 (TNF) 在缺陷菌株中的表达量占到全菌蛋白的 20.8%~24.3%,说明ptsG基因缺陷株具有良好的生长能力和表达外源蛋白的能力,在大肠杆菌高密度发酵[11-13]研究方面具有巨大的应用前景。
现有的一些发酵菌种可利用碳源的范围比较窄 (如运动发酵单胞菌只能以葡萄糖、蔗糖或果糖作为发酵原料;酿酒酵母也只能利用六碳糖,不能利用五碳糖),而大肠杆菌虽然可利用的碳源范围较广 (可以利用五碳糖和六碳糖),但是在五碳糖和六碳糖同时存在的情况下,由于碳代谢产物抑制,大肠杆菌是先利用完葡萄糖,再利用木糖的,木糖的消耗量不大。为了提高糖的消耗量,提高乙醇产量,文中采用遗传背景清楚、易操作、易调控、培养基要求简单和生长迅速的工程菌大肠杆菌SZ470为菌株,利用Red同源重组技术[14]构建ptsG基因缺陷株,有望降低葡萄糖的摄取速率,减少副产物累积,解除葡萄糖效应,促进菌体生长,使其在混合糖 (即五碳糖和六碳糖) 培养基中能同时利用五碳糖和六碳糖,扩大大肠杆菌底物同时利用糖的范围,增加糖的消耗量,提高乙醇的产量。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
Escherichia coliSZ470为本实验室构建的高效同型乙醇发酵工程株,来源于野生菌株E. coliB,其丙酮酸甲酸裂解酶 (focA-p fl B)、延胡索酸还原酶 (frdABCD)、乙酸激酶 (ackA)、D-乳酸脱氢酶 (ldhA) 4个基因被敲除,并通过无氧启动子融合表达技术倍增了 NADH还原力,是一株优秀的厌氧同型乙醇发酵菌株[15-17],该菌株由于敲除了ldhA、frdABCD、p fl B、ackA等基因,因此减少或消除了其他副产物,使其葡萄糖的代谢产物主要只有乙醇。质粒pKD46 (温度敏感型复制子,Ampr)、质粒pKD4 (含有卡那霉素的抗性基因Kanr) 和抗性基因消除质粒 pCP20为本实验室保存[18]。
1.1.2 引物
扩增卡那霉素抗性基因的引物P1、P2分别由两部分组成 (表1),靠近5端未加下划线的序列与ptsG基因两翼序列同源,靠近 3端加下划线的序列与质粒pKD4上Kanr基因两侧序列互补。采用 Red同源重组技术[14](它是一段两侧40~60 bp,与目标基因同源的序列,利用Red重组酶,将它导入菌体细胞中,使扩增的DNA片段与染色体的特定目标序列进行同源重组,目标基因被标记基因替换下来的一种基因工程敲除手段) 使其与目标基因互换。在大肠杆菌染色体上ptsG基因外侧和内侧,分别设计两对引物P3、P4和P5、P6用于敲除后重组菌株的验证。引物合成由上海生工生物工程技术服务有限公司完成。
1.1.3 主要试剂和仪器
DNATaq聚合酶购自Fermentas公司。L-阿拉伯糖为国药集团化学试剂有限公司产品。蛋白胨和酵母粉为英国Oxoid公司产品。氨苄青霉素和卡那霉素均购自 Mersco公司,工作浓度分别为100 mg/L和50 mg/L。其他化学试剂均购自国药集团化学试剂有限公司。电击转化仪为美国Bio-Rad公司生产。
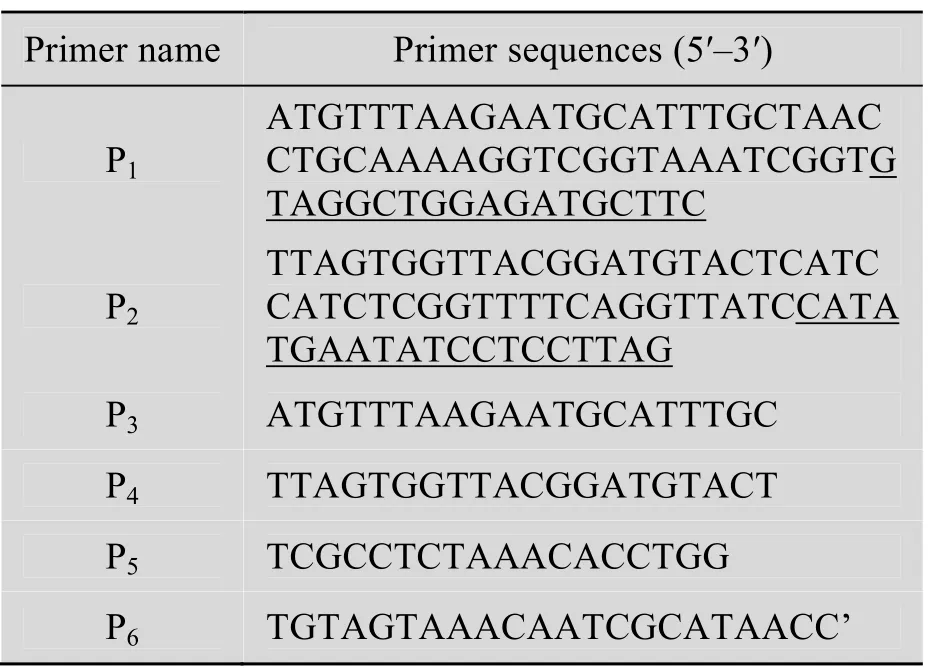
表1 敲除和鉴定ptsG基因的引物Table 1 Primers for deletion and identification of ptsG
1.1.4 培养基
LB培养基:蛋白胨10 g/L、酵母粉5 g/L、氯化钠5 g/L;发酵培养基:LB培养基加5%的葡萄糖 (LB+5% G)、LB培养基加2.5%的葡萄糖和2.5%的木糖 (LB+2.5% G+2.5% X,即混合培养基)。
1.2 方法
1.2.1 PCR片段的制备
以质粒 pKD4为模板,P1和 P2为引物,进行PCR扩增,反应条件为:95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸2 min,循环30次。PCR产物经回收纯化后,溶于去离子水中,得到两端带有ptsG同源臂的卡那霉素抗性基因片段。
1.2.2 pts G基因敲除
用CaCl2法将pKD46转化到E. coliSZ470细胞中,经过氨苄青霉素抗性平板筛选后得到阳性菌落。挑取大肠杆菌SZ470/pKD46的单菌落接种于新鲜LB培养基中,加入2% L-阿拉伯糖30 ℃培养至OD600=0.5~0.6,将菌液置冰浴中 15 min后10 000 r/min离心10 min,所得细胞用去离子水反复洗涤3次后。将上述PCR扩增产物与EP管中的细胞混合,1 500 V电击后立刻加入1 mL预冷的LB+50% G的培养基中,于摇床中30 ℃、150 r/min复苏培养2 h,涂布于含有卡那霉素的抗性平板上,37 ℃恒温培养24 h,筛选阳性转化子。
1.2.3 重组菌株的鉴定及抗性基因的消除
挑取在卡那霉素抗性平板上生长良好的阳性转化子单菌落,以 P3、P4和 P5、P6为引物进行 PCR扩增,并通过琼脂糖凝胶电泳检测 PCR产物大小,以E. coliSZ470为对照菌株。将pCP20质粒转化到已成功敲除ptsG基因的卡那霉素抗性阳性菌株,30 ℃培养3 h后,涂于无抗性平板上,42 ℃过夜培养,热诱导促进FLP重组酶表达,同时质粒也逐渐丢失。挑取在无抗性平板上生长良好而在抗性平板上不能生长的重组菌菌落,转接3次。
1.2.4 五碳糖发酵培养
挑取敲除成功的重组菌株、对照菌株的单菌落接种于装有100 mL LB培养基的250 mL的三角瓶中,37 ℃培养 10 h后,分别接种于装有100 mL LB+5% G和LB+2.5% G+2.5% X培养基的250 mL三角瓶中,37 ℃、150 r/min继续培养,定时取样,测定菌体光密度OD600值、糖浓度及乙醇产量。每个处理做3个平行。
1.2.5 发酵产物检测分析
光密度OD600值用紫外分光光度计在600 nm处测定,葡萄糖和木糖的浓度用安捷伦的1 200型高效液相色谱仪检测,检测条件:采用示差检测器,柱子型号为 BioRad公司的 HPX87H,柱温 35 ℃,流动相为 4 mmol/L H2SO4,流速为0.5 mL/min,进样 20 μL;乙醇采用气相色谱仪进行测定,测定条件:采用氢离子火焰检测器(FID),毛细管柱,柱温40 ℃,进样温度200 ℃,检测器温度200 ℃,载气为氮气,流速2 mL/min,正丙醇作为内标,进样0.5 μL。乙醇理论产量的计算为消耗1 mol葡萄糖可产生2 mol乙醇,消耗1.2 mol木糖可产生2 mol乙醇。根据消耗的糖的量来计算实际产乙醇的量。
2 结果
2.1 PCR片段的制备
以 pKD4为模板,P1和 P2为引物,扩增出两端与ptsG基因上下游序列同源,中间为卡那霉素抗性基因Kanr的 DNA片段,经 1.0%琼脂糖凝胶电泳检测,片段大小约为1 550 bp,与理论值基本一致 (图1)。
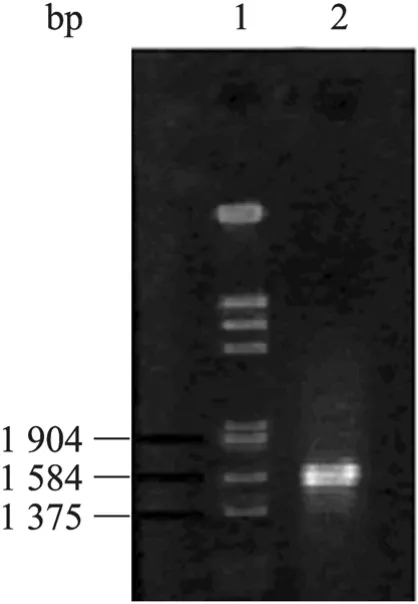
图1 PCR产物的琼脂糖凝胶电泳分析Fig. 1 Electrophoresis analysis of PCR product. 1:DNA marker; 2: PCR product.
2.2 ptsG基因缺陷株的鉴定
挑取卡那平板上筛选得到的转化菌株单菌落,以P3和P4为引物,进行PCR扩增,电泳检测PCR产物大小。结果如图2A所示,ptsG基因自身长度为1 434 bp,电击转化后,卡那霉素抗性基因重组到SZ470染色体上,扩增的片段长度约为1 550 bp,而两者相差100 bp左右;以P5、P6为引物,按上述步骤进行再 PCR扩增,由于引物 P5、P6是在ptsG基因内部设计的,所以如果敲除成功,重组菌株不会出现相应的扩增条带,而对照菌株会出现约786 bp大小的基因片段(图2B)。通过两次PCR验证实验,可以证明ptsG基因成功敲除,将敲除成功的菌落经过42 ℃热诱导培养,使其卡那霉素抗性基因消除掉,将该鉴定成功敲除ptsG基因的重组菌株命名为Escherichia coliSZ470P,用于后续摇瓶发酵实验,以进一步验证预期的结果。
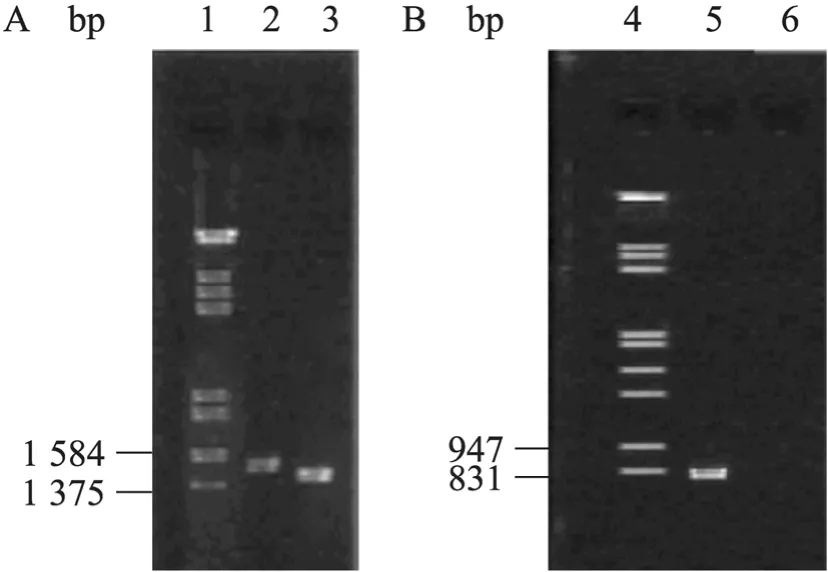
图2 ptsG基因缺陷株的PCR鉴定Fig. 2 Identification of ptsG mutants by PCR. (A) PCR was performed with the primers P3 and P4. (B) PCR was performed with the primers P5 and P6. 1, 4: DNA marker; 2, 6: SZ470P; 3, 5: SZ470.
2.3 重组菌株生长曲线
以在培养基中各时刻所测定的菌体吸光度对发酵时间作图,从图3可以看出,在LB+5% G的培养基中,ptsG缺陷菌株 SZ470P在发酵0~48 h,缺陷菌株OD600值小于对照组;发酵48 h后,缺陷菌株 SZ470P的OD600值超过了对照菌株,在72 h达到最大OD600值为10.65,是对照菌株的1.15倍,对照菌株最高OD600值为9.24。
在LB+2.5% G+2.5% X培养基中,ptsG缺陷菌株最大OD600值 (9.57) 稍高于对照菌株最大OD600值 (9.27)。以上结果表明,与对照菌株相比,大肠杆菌ptsG缺陷株可摄取更多的还原糖用于菌体生长,因而具有较高的生物量。
2.4 重组菌株还原糖消耗曲线
由于敲除了ptsG基因,ptsG基因缺陷菌株SZ470P的葡萄糖消耗量就不及对照菌株SZ470,如图4A所示,在LB+5% G的培养基中,缺陷菌株SZ470P葡萄糖的消耗速率明显低于对照菌株,其葡萄糖消耗速率 (1.08 g/(L·h)) 是对照菌株 (2.08 g/(L·h)) 的一半。发酵到最后,对照菌株消耗了39 g/L的葡萄糖,而缺陷菌株只消耗了大约40%的葡萄糖,即消耗了20 g/L。
从图4B中可以看到,对照菌株SZ470是先利用完葡萄糖再利用木糖的,即在发酵36 h时消耗完葡萄糖之后,再开始消耗木糖,葡萄糖的消耗速率达到了1 g/(L·h),而木糖的消耗速率仅为0.17 g/(L·h),这是因为葡萄糖和木糖同时存在的情况下,葡萄糖会产生抑制效应。而缺陷菌株SZ470P培养基中的木糖和葡萄糖的消耗曲线是同时下降的,其中木糖的消耗速率 (0.64 g/(L·h))比葡萄糖 (0.5 g/(L·h)) 的快。如图4B所示,发酵到96 h时,对照菌株葡萄糖全部消耗完,木糖只消耗了 5 g/L;而缺陷菌株 SZ470P消耗了13 g/L的葡萄糖和 20 g/L的木糖。缺陷菌株SZ470P的木糖消耗量是对照菌株的 4倍。可能是因为敲除了ptsG基因之后,大肠杆菌减少了对葡萄糖的摄取率,减弱了葡萄糖抑制效应,使大肠杆菌能同时利用葡萄糖和木糖,增加了木糖的消耗量。
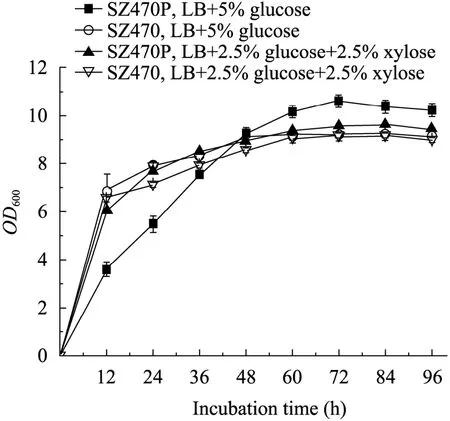
图3 大肠杆菌ptsG缺陷株在LB+5% 葡萄糖及混合糖培养基中的生长情况Fig. 3 Comparison of growth between parent and ptsG mutant strains in LB+5% glucose and sugar mixture medium.
2.5 乙醇产量
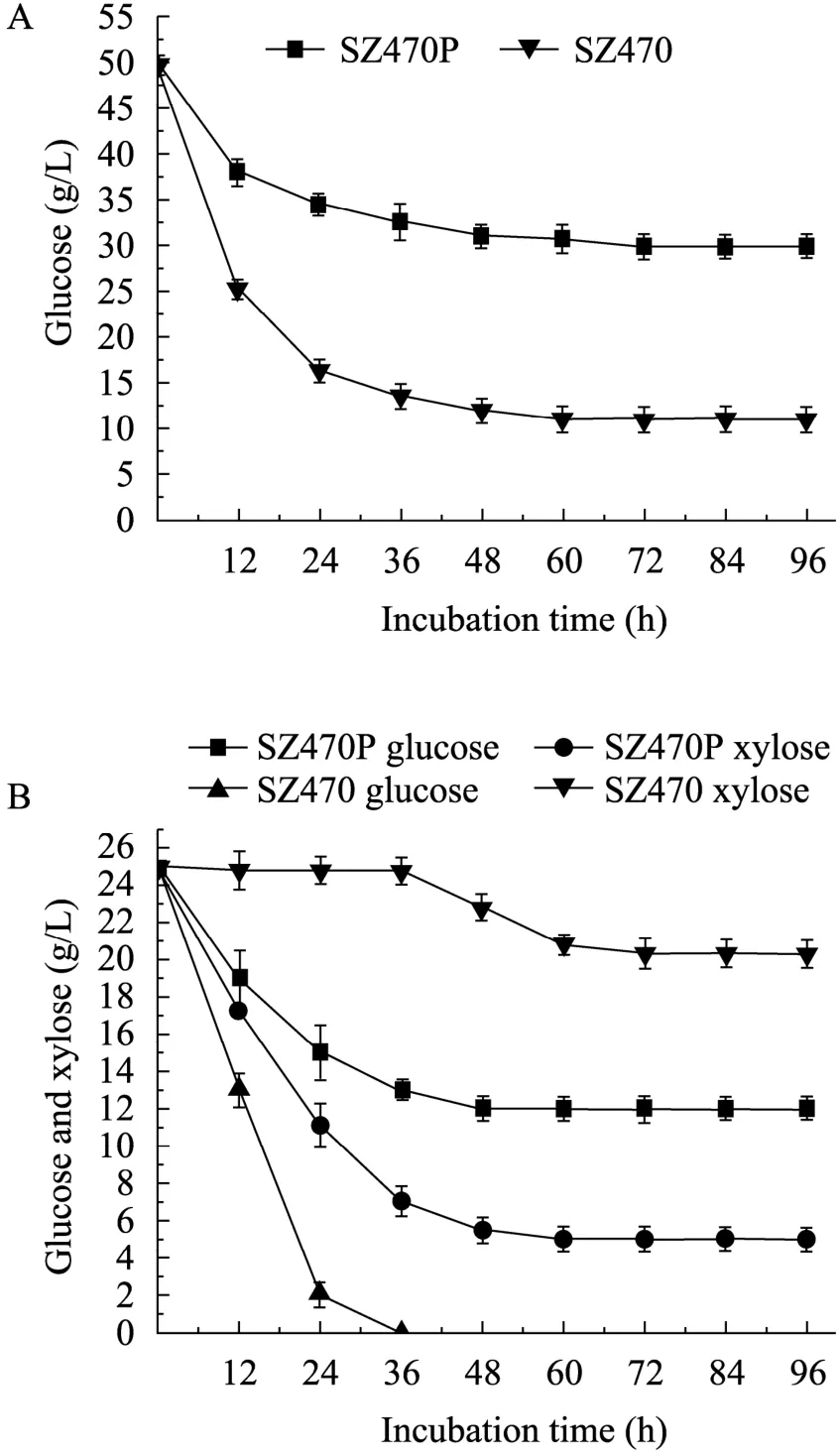
图4 大肠杆菌ptsG缺陷株和对照菌株对葡萄糖和木糖的利用Fig. 4 Sugar consumption of E. coli SZ470P (ptsG−)and E. coli SZ470 (ptsG+). (A) Fermentation in 5%glucose. (B) Fermentation in 2.5% glucose+2.5%xylose.
大肠杆菌缺陷菌株SZ470P与对照组SZ470的发酵乙醇产量如图 5所示,缺陷菌株 SZ470P在LB+5% G的培养基中的乙醇产量不高,因为其被敲除了ptsG基因之后发酵前期生长比较缓慢,利用的葡萄糖量减少,在发酵48 h时,乙醇产量只有7.3 g/L,在发酵84 h才达到最高乙醇产量 11.16 g/L,大约是对照菌株最高产量(19.69 g/L) 的一半;说明ptsG基因对控制葡萄糖转运有很大影响。
在LB+2.5% G+2.5% X培养基中,在发酵12 h时,缺陷菌株与对照组的乙醇产量分别为7.42 g/L和10.74 g/L,在发酵前24 h,缺陷菌株的乙醇产量没有对照菌株的高,而在发酵 24 h之后,缺陷菌株的乙醇产量超过了对照组,由原来对照组的最高乙醇产量 13.13 g/L提高到了15.01 g/L,提高了14.32%;缺陷株SZ470乙醇转化率为理论值的 89.13%。这可能是因为SZ470P能同时利用已糖 (葡萄糖) 和戊糖 (木糖),从整体上提高了还原糖的消耗量,扩大了底物同时利用糖的范围,提高了糖的转化率,最终提高了乙醇的产量。
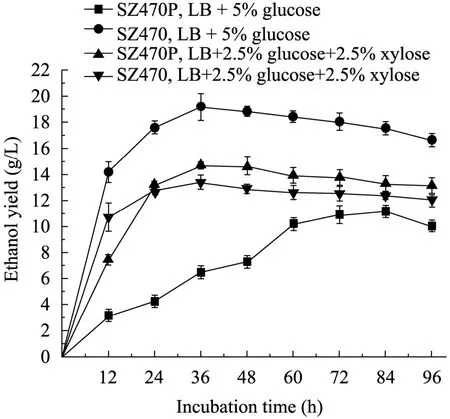
图5 大肠杆菌ptsG缺陷株和对照菌株的乙醇产量Fig. 5 Ethanol yield of ptsG mutant strains and parent strains.
3 讨论
利用基因打靶技术敲除生物代谢途径中的关键基因,改变代谢流分布,构建新的代谢途径,在微生物代谢工程研究中具有重要作用。本文利用Red同源重组技术敲除大肠杆菌的ptsG基因,在 LB+5% G培养基中,在发酵前 48 h,ptsG基因缺陷菌株的OD600值低于亲株,生长比较缓慢[19],是因为敲除了指导菌体将胞外的葡萄糖转运到胞内的主要基因ptsG,使葡萄糖从胞外转运到胞内的速度减慢所致。基因缺陷株的菌密度在发酵48 h之后,明显比亲株高,在进入对数生长期后获得最大OD600值为 10.65,比对照组的最大菌密度高出11.2%。敲除后的SZ470P达到最大菌体密度的时间比对照菌株延后了,因为在大肠杆菌转运葡萄糖的系统中,除了磷酸转移酶系统 (PTS) 之外,还有一些其他酶 (如酶ⅡGlc等)均可将葡萄糖转运入微生物细胞内[3],因此,敲除基因ptsG后细胞可通过其他转运酶的作用将胞外的葡萄糖转运到胞内,来摄取葡萄糖,在生长过程中摄取葡萄糖用于菌体的生长和繁殖,其最高OD600值是对照菌株的1.5倍。敲除了ptsG基因之后,大肠杆菌减少了对葡萄糖的摄取率,其葡萄糖的消耗量是对照菌株的50%,减弱了葡萄糖抑制效应,使大肠杆菌葡萄糖代谢产物抑制现象得以解除,在六碳糖 (葡萄糖) 和五碳糖(木糖、阿拉伯糖等) 同时存在的情况下,缺陷株SZ470P能同时利用五碳糖和六碳糖,由于葡萄糖的抑制效应得以解除,从而使木糖的消耗量由原来的 20%提高到 80%。在只有葡萄糖存在的培养基中,缺陷株乙醇产量是对照菌株的一半,这一方面是由于SZ470P菌株被敲除了转运葡萄糖的ptsG基因,使消耗的葡萄糖减少;另一方面,摄取的葡萄糖更多的转化为菌体的生物量。而在六碳糖 (葡萄糖) 和五碳糖 (木糖) 同时存在的情况下,混合糖能被同时利用,还原糖的消耗量提高,从而使其转化成乙醇的量也相应提高。
目前,在利用原料上,特别是以秸秆为廉价原料[1,20-23],稀酸水解之后进行发酵[24],其水解液中同时有五碳糖 (如木糖、阿拉伯糖等) 和六碳糖 (如葡萄糖、半乳糖、甘露糖等),如果利用酵母来进行发酵,则会造成大量五碳糖的浪费。利用代谢工程技术,构建ptsG缺陷的大肠杆菌基因工程菌,可大大提高还原糖的利用,扩大同时利用底物的范围,降低生产成本。由于高密度发酵过程的重要性[25],大肠杆菌ptsG基因缺陷株的发酵特性及乙醇产量的提高还有待在发酵罐中进一步研究,在实现缺陷菌株高密度生长的同时,提高乙醇的产量。
[1]Li R, Chen Q, Wang PG, et al. A novel-designedEscherichia colifor the production of various polyhydroxyalkanoates from inexpensive substrate mixture.Appl Microbiol Biotechnol, 2007, 75(5):1103−1109.
[2]Stulke J, Hille W. Carbon catabolite repression in bacteria.Curr Opin Microbiol, 1999, 2(2):195−201.
[3]Bruckner R, Titgemeyer F. Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization.Microbiol Lett, 2002, 209(2): 141−148.
[4]Jackson AU, Werner SR, Talaty N, et al. Targeted metabolomic analysis ofEscherichia coliby desorption electrospray ionization and extractive electrospray ionization mass spectrometry.Anal Biochem, 2008, 375(2): 272–281.
[5]Tanaka Y, Teramoto H, Inui M, et al. Regulation of expression of general components of the phosphoenolpyruvate: carbohydrate phosphotransferase system (PTS) by the global regulator suger in corynebacterium glutamicum.Appl Microbiol Biotechnol, 2008, 78(2): 309−318.
[6]Deutscher J, Francke C, Postma PW. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria.Microbiol Mol Biol R, 2006, 70(4):939−1031.
[7]Zhou JZ, Zou YK, Dai HM, et al. One-step knockout of theptsHIcrroperon inEscherichia coliand characterization of the mutant. Microbiol China, 2010, 37(8): 1146−1152 (in Chinese).
周军智, 邹永康, 戴红梅, 等. 大肠杆菌pts HIcrr操纵子的快速敲除及敲除菌生长性能测定.微生物学通报, 2010, 37(8): 1146−1152.
[8]Siebold C, Flukiger K,Beutler R. Carbohydrate transporters of the bacterial phosphoenolpyruvate:sugar phosphotransferase system (PTS).FEBS Lett,2001, 504(3): 104−111.
[9]Chou CH, Bennett GN, San KY. Effect of modified glucose uptake using genetic engineering techniques on high-level recombinant protein productionEscherichia colidense cultures.Biotechnol Bioeng, 1994, 44(8): 952−960.
[10]Han C, Zhang WC, You S, et al. Knockout of theptsGgene inEscherichia coliand cultural characterization of the mutants. Chin J Biotech,2004, 20(1): 16−20 (in Chinese).
韩聪, 张惟材, 游松, 等. 大肠杆菌ptsG基因敲除及其缺陷株生长特性研究. 生物工程学报,2004, 20(1): 16−20.
[11]Xiao XX, Han MJ, Yoo JS. Comparison of the extracellular proteomes ofEscherichia coliB and K-12 strains during high cell density cultivation.J Proteomics, 2008, 8(10): 2089−2103.
[12]Soini J, Ukkonen K, Neubauer P. High cell density media forEscherichia coliare generally designed for aerobic cultivationsconsequences for large-scale bioprocesses and shake flask cultures.Microb Cell Fact, 2008, 7(1): 26.
[13]Krause M, Ukkonen K, Haataja T, et al. A novel fed-batch based cultivation method provides high cell-density and improves yield of soluble recombinant proteins in shaken cultures.Microb Cell Fact, 2010, 9(1): 11.
[14]Zhang Q, Gao HJ, Dong MY. Advances in red recombination applied in knockout of bacteria chromosomal gene. China Biotechnol, 2006, 26(1):81−86 (in Chinese).
张全, 高会杰, 佟明友. Red 重组技术研究进展.中国生物工程杂志, 2006, 26(1): 81−86.
[15]Chen K, Iverson AG, Garza EA, et al. Metabolic evolution of non-transgenicEscherichia coliSZ420 for enhanced homoethanol fermentation from xylose.Biotechnol Lett, 2010, 32(1): 87−96.
[16]Zhou S, Iverson A, Grayburn WS. Engineering a native homoethanol pathway inEscherichia coliB for ethanol production.Biotechnol Lett, 2008,30(2): 335−342.
[17]Zhou S, Iverson AG, Grayburn WS. Doubling the catabolic reducing power (NADH) output ofEscherichia colifermentation for production of reduced products.Biotechnol Progr, 2010, 26(1):45−51.
[18]Datsenko K, Wanner B. One-step inactivation of chromosomal genes inEscherichia coliK-12 using PCR products.Proc Natl Acad Sci USA, 2000,97(12): 6640.
[19]Nichols N, Dien B, Bothast R. Use of catabolite repression mutants for fermentation of sugar mixtures to ethanol.Appl Microbiol Biotechnol,2001, 56(1): 120−125.
[20]Saha BC, Cotta MA. Ethanol production from alkaline peroxide pretreated enzymatically saccharified wheat straw.Biotechnol Progr, 2008,22(2): 449−453.
[21]Ko JK, Bak JS, Jung MW, et al. Ethanol production from rice straw using optimized aqueous ammonia soaking pretreatment and simultaneous saccharification and fermentation processes.Bioresource Technol, 2009, 100(19): 4374−4380.
[22]Huang CF, Lin TH, Guo GL, et al. Enhanced ethanol production by fermentation of rice straw hydrolysate without detoxification using a newly adapted strain ofPichia stipitis.Bioresource Technol, 2009, 100(17): 3914−3920.
[23]Bak JS, Ko JK, Choi IG, et al. Fungal pretreatment of lignocellulose byPhanerochaete chrysosporiumto produce ethanol from rice straw.Biotechnol Bioeng, 2009, 104(3): 471−482.
[24]Sun Y, Cheng JJ. Dilute acid pretreatment of rye straw and bermudagrass for ethanol production.Bioresource Technol, 2005, 96(14): 1599−1606.
[25]Lu YH, Knol J, Flaschel E, et al. Cultivation of immobilizedDictyostelium discoideumfor the production of soluble human fas ligand.Appl Microbiol Biotechnol, 2004, 65(5): 547−552.
