毛皮中6种致病菌基因芯片检测方法的建立和应用
2013-08-30冯松凯李苗云刘胜男吕晓飞
冯松凯,李苗云,徐 超,张 勤,苗 丽,李 珂,杨 娜,刘胜男,吕晓飞
(1.河南农业大学食品科学技术学院,河南郑州 450002;2.河南出入境检验检疫局,河南郑州 450003;3.三门峡出入境检验检疫局,河南三门峡 472000)
近年来,我国已经成为世界上最大的皮毛进口国,进口皮张来源广泛。由于动物皮毛是细菌的良好培养基,其携带致病菌的风险越来越引起人们的关注。入境皮毛中携带的致病菌主要为布氏杆菌(Brucella)、大肠杆菌O157(E.coliO157)、金黄色葡萄球菌(Staphylococcus aureus)、乙型溶血性链球菌(β-streptococcus)、 丹 毒 杆 菌 (Erysipelothrix rhusiopathiae)、铜绿假单胞杆菌(Pseudomonas aeruginosa)等。其中Brucella、E.coliO157和S.aureus等感染人的事件已有报道[1-3]。目前,国内外对皮毛携带致病菌的检测方法研究较少,而主要依靠经典的细菌学鉴定和PCR法[4-6],缺少对皮毛携带致病菌的高通量检测方法。因此,本研究选择在细菌分类鉴定学上具有重要生物学意义的16S rDNA基因和gyrB基因进行序列分析并设计两对通用引物和相应的多条细菌鉴别探针,研究同时检测6种致病菌的基因芯片检测方法,并进行临床应用效果评价。
1 材料和方法
1.1 菌 株B rucella、S.aureus26003-5a7株、β-streptococcus32210-4a株、E.rhusiopathiaeCVCC123、蜡样芽孢杆菌(B.cereus)CVCC2002和B.cereusCVCC2003均购自国家兽医微生物菌种保藏中心;P.aeruginosaATCC15442和P.aeruginosaATCC10145由山东出入境检验检疫局惠赠;苏云金杆菌(B.thuringiensis)S1株和B.thuringiensisS6株由中国军事医学科学院微生物传染病所惠赠;E.coliO157 NCTC 12900、S.aureusATCC29213、E.coliO152、李斯特杆菌(Listeria)、沙门氏菌(Salmonella)和阪崎肠杆菌(E.sakazakii)均由本实验室鉴定保存。
1.2 主要试剂及仪器 细菌基因组DNA提取试剂盒购自北京明日百傲生物科技发展研究所;MasterM ixTaq酶、100 bp DNA Marker均购自天跟生化科技有限公司;晶芯R基因芯片点样液、光学级氨基基片购自博奥生物有限公司;SYBRRPremix ExTaqTM试剂盒购自TaKaRa公司;Smart ArrayerTM48芯片点样仪、BioM ixerTMⅡ芯片杂交仪、晶芯SlideWasher芯片洗干仪、晶芯LuxScanIM 10K芯片扫描仪为博奥生物有限公司产品;LabCycler Standard梯度PCR仪为SENSO公司产品。
1.3 探针和引物的设计与合成 选择细菌的16S rDNA基因和gyrB基因为靶基因,分别设计通用引物和每种致病菌的特异性寡核苷酸探针。上游引物5'端均标记TAMARA荧光基团,探针碱基长度小于35 bp的探针5'端加T至35 bp(表1和表2)。
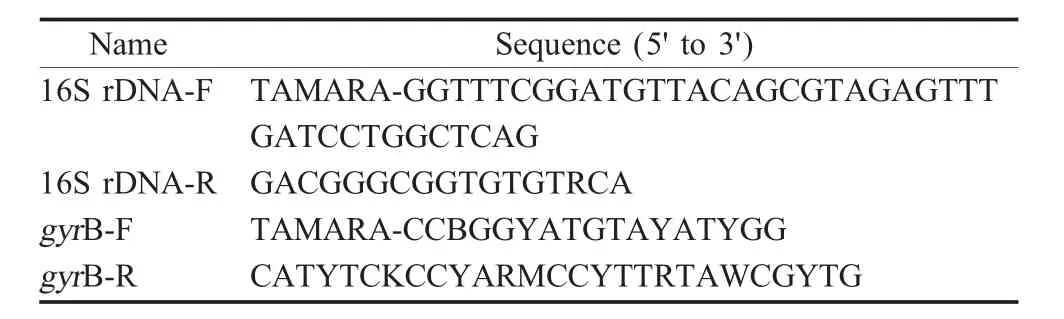
表1 基因芯片检测引物序列Table 1 The sequence of primers
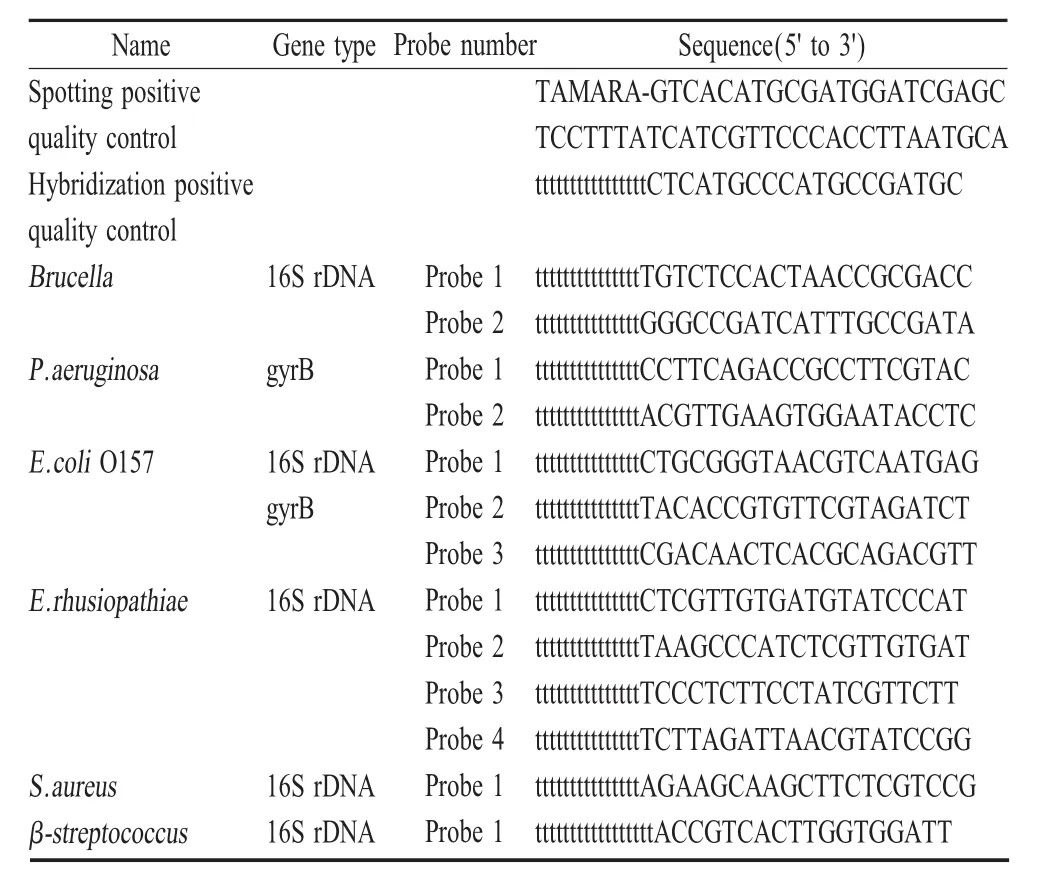
表2 基因芯片检测探针序列Table 2 The sequence of probes
1.4 细菌DNA的提取 无菌采集进境动物皮毛,接种LB液体培养基进行富集。取菌液8 000 r/m in离心5m in,采用细菌核酸提取试剂盒提取其DNA。同时,提取参考菌株DNA备用。
1.5 基因芯片检测方法的建立和优化
1.5.1 基因芯片的制备以晶芯R基因芯片点样液稀释各探针至终浓度30μmol/L,将各探针阵列点样于空白基片。每张基片设12个矩阵,每个矩阵为18×10阵列(图1)。点样完毕,将制备的芯片80℃固定探针2 h~3 h后,4℃保存备用。
1.5.2 不对称PCR方法的建立采用S.aureus、E.rhusiopathiae、Brucella、β-streptococcus、E.coliO157、P.aeruginosa6种致病菌基因组DNA,并以其为模板建立基于16S rDNA基因的不对称PCR方法,优化退火温度为57℃;同时,以E.coliO157和P.aeruginosa基因组DNA为模板,建立基于gyrB基因的不对称PCR方法,优化退火温度为55℃。两种方法不对称PCR反应体系为:2×TaqPCR MasterM ix 12.5μL,上游引物(20μmol/L)5.0μL,下游引物(20μmol/L)0.5μL,模板2.0μL,补ddH2O至25.0μL;不对称PCR反应条件为:94℃ 5 Min;94℃ 1m in、57℃或55℃ 1 Min、72℃ 1 min,共30个循环;72℃10m in。

图1 基因芯片检测探针布局Fig.1 The position of detection probes for DNA chip
1.5.3 基因芯片的杂交与洗涤取8μL 42℃预热的杂交液a(47%甲酰胺、7×SSC、0.4%SDS、9×Denbardt's)与7μL PCR产物混匀,95℃变性10m in,迅速冰浴5 Min。参照晶芯R基因芯片杂交盒说明,将杂交样品点加到基因芯片每个阵列中。芯片杂交仪中42℃、5 r/m in摇转杂交4 h。以洗液Ⅰ(2×SSC、0.2%SDS)42℃振荡洗涤3 min,重复3次;洗液Ⅱ(0.2×SSC)洗涤3次;超纯水清洗2次后,300 r/m in离心甩干后备检。
1.5.4 基因芯片扫描与分析预热芯片扫描仪,选择Cy3通道扫描杂交的基因芯片,LuxCan3.0软件记录、分析荧光信号强度。
1.5.5 基因芯片杂交条件的优化选择不同甲酰胺浓度的4种杂交液,参照1.5.3方法以致病菌PCR产物与制备的基因芯片分别30 Min、1 h、2 h、4 h杂交过夜,扫描、记录杂交信号,根据杂交信号的强度选择最佳杂交条件。杂交液配方为:杂交液a、b(3×SSC、0.2%SDS、25%甲酰胺、5×Denhardt's)、c(5×SSC、2.5%甲酸胺、0.2%SDS)和 d(20×SSC 1.5μL,10%SDS 0.25μL)。
1.6 特异性试验 用溶液型细菌基因组DNA提取试剂盒分别提取12种致病菌核酸,采用不对称PCR方法对其进行扩增,与制备基因芯片杂交。
1.7 敏感性试验 分别测定S.aureus、E.rhusiopathiae、Brucella、β-streptococcus、E.coliO157、P.aeruginosa6种致病菌核酸浓度,用ddH2O 10倍梯度稀释,共做6个稀释度,进行不对称PCR扩增,与制备的基因芯片杂交进行敏感性检测。
1.8 保存期试验 以S.aureus、E.rhusiopathiae和BrucellaPCR产物分别与4℃避光1个月、2个月、4个月、6个月的基因芯片杂交进行基因芯片的稳定性检测。
1.9 方法比较 随机选取163个送检的皮毛样品,分别称取2 g,无菌剪碎后,加入5 ML LB培养液,37℃震荡培养2 h~6 h。将菌液8 000 r/m in离心5 min沉淀菌体,提取核酸,进行芯片杂交。同时参照文献[7]中PCR方法进行Brucella的鉴定及文献[8]中荧光定量PCR方法进行S.aureus的鉴定。
2 结 果
2.1 不对称PCR扩增结果 采用16S rDNA通用引物进行PCR扩增,经琼脂糖凝胶电泳检测,结果表明S.aureus、E.rhusiopathiae、Brucella、β-streptococcus、E.coliO157、P.aeruginosa6种致病菌均扩增出约1.4 kb的目的条带;而采用gyrB基因通用引物对E.coliO157和P.aeruginosa核酸进行PCR扩增,均扩增出约2.0 kb的目的条带,与预期片段相符。
2.2 基因芯片杂交条件优化结果 分别选择4种不同的杂交液和不同的杂交时间,参照1.5.3方法以S.aureusPCR产物与制备的基因芯片杂交,根据杂交信号的强度,结果显示:选择杂交液a,在42℃、5 r/m in条件下杂交4 h杂交信号最强。
2.3 特异性试验 以提取的12种致病菌核酸为模板,PCR扩增后,每种致病菌的扩增产物分别与制备基因芯片杂交。结果显示扩增的Brucella、E.coliO157、S.aureus、β-streptococcus、E.rhusiopathiae、P.aeruginosa产物在相应检测探针位点出现绿色荧光信 号 , 扩 增 的B.cereus、E.coliO152、Listeria、Salmonella、E.sakazakii、B.thuringiensis产物 与基 因芯片杂交未出现信号(表3)。表明各致病菌探针间无交叉反应,制备的基因芯片具有很强的特异性。
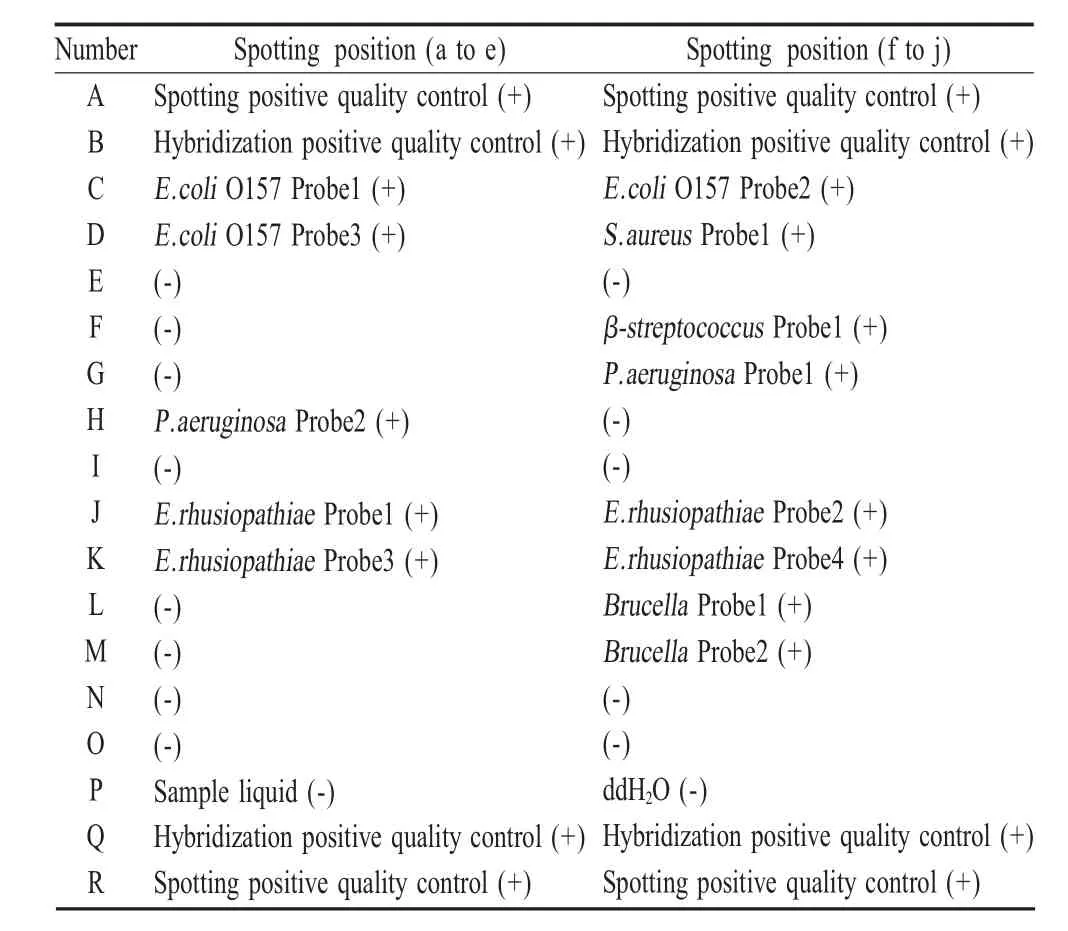
表3 基因芯片特异性检测结果Table 3 The specific result of DNA chip
2.4 敏感性试验 分别提取Brucella、E.coliO157、S.aureus、β-streptococcus、E.rhusiopathiae、P.aeruginosa的基因组DNA,以10倍梯度稀释,共做6个稀释度,进行PCR扩增后与基因芯片进行杂交。结果表明:基因芯片可检测到Brucella、E.coliO157、S.aureus、β-streptococcus、E.rhusiopathiae、P.aeruginosa阳性杂交信号核酸最低浓度分别约为10拷贝、10拷贝、100拷贝、100拷贝、10拷贝、100拷贝。
2.5 保存期试验 与4℃避光保存的基因芯片进行杂交,结果显示3种细菌的PCR产物与保存至少6个月的基因芯片杂交仍能出现阳性信号。此外,将制备的芯片在室温条件下避光保存3、5和6个月,检测结果显示芯片仍具有很强的荧光信号。
2.6 方法比较结果 对选取的163张皮毛样品进行基因芯片杂交,样品1号和23号检测结果为Brucella阳性(图2),同时以Brucella引物进行PCR方法对皮毛进行检测,结果1号和23号样品均扩增出单一目的条带(图3)。样品32号芯片检测结果为S.aureus阳性(图 2)。采用S.aureus实时荧光定量PCR方法对样品32号进行检测,可扩增出特异性目的曲线(图4),与PCR鉴定结果一致。对163份皮毛样品检测结果与PCR检测结果符合率为100%。

图2 基因芯片对皮毛样品检测结果Fig.2 Fur sample test results by DNA chip
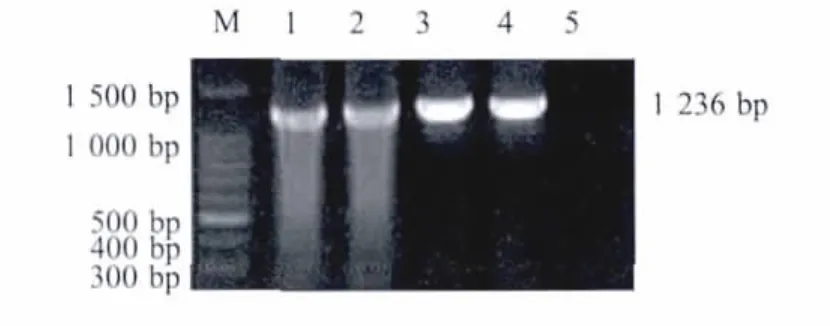
图3 Brucella引物对1号和23号皮毛样品PCR检测结果Fig.3 The PCR detections of fur sample No.1 and No.23 w ith Brucella specific primer
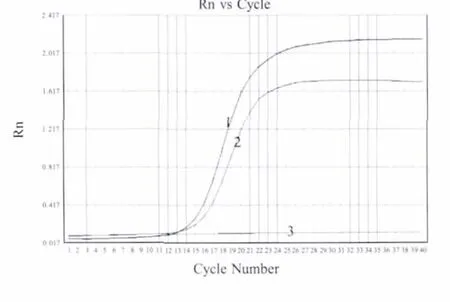
图4 S.aureus引物对32号皮毛样品荧光定量PCR检测结果Fig.4 The fluorescence quantitative PCR curve of fur sample No.32 with S.aureus specific primer
3 讨 论
目前,对皮毛中携带致病菌检测依然使用传统的方法,难以实现高通量检测以及对携带的致病菌的快速筛选。本研究采用荧光标记设计的两对通用引物,采用新的不对称PCR技术,扩增后产生大量单链标记核酸片段,在此基础上建立皮毛中6种致病菌的基因芯片检测技术。该方法特异性强,灵敏度高,可以实现高通量快速检测,适合现代进出境检验检疫部门批量样品检测的需求。
影响基因芯片检测技术关键因素之一是探针的设计和选择。特异性寡核苷酸探针片段在致病菌核酸中的位置、探针的长度以及Tm值均影响芯片检测的特异性、敏感性和杂交信号的强度[9-10]。本研究在设计探针时,充分考虑了寡核苷酸探针的Tm值,以保证在同一杂交温度下基因芯片对样品进行高通量检测。同时,考虑到探针在芯片上的空间占位效应,研究将设计探针长度短于35 bp的在其5'端增加T碱基至35 bp。试验结果表明,该方法设计的探针特异性好,荧光信号强于未加T碱基的其它探针。在基因芯片杂交过程中,杂交液中甲酰胺浓度、杂交温度以及杂交时间共同影响着基因芯片杂交的特异性和敏感性。不适当的杂交条件影响芯片杂交信号的强度,甚至出现假阳性杂交信号[11]。本研究选择4种不同浓度甲酰胺杂交液,同时对杂交温度以及杂交时间进行优化,最终确定基因芯片的最佳杂交条件,检测最高灵敏度达到约10个基因拷贝数。
尽管基因芯片检测技术因其高通量、微型化以及自动化等方面具有优越性[12-14],但是作为一种新技术还存在亟待解决的问题,如建立标准化程序、降低检测费用以及进一步提高检测的特异性等。本研究建立的方法能够有效的检测动物皮毛中携带的6种致病菌,同时该项技术为环境以及其他临床样品中致病菌的污染和传播检测提供了技术平台。
[1]毛景东,王景龙,杨艳玲.布鲁氏菌病的研究进展[J].中国畜牧兽医,2011,38(1):222-227.
[2]孟祥升,辛崇兴,邵晞.肠出血性大肠杆菌O157∶H7研究进展[J].中国动物检疫,2011,28(11):69-71.
[3]邓燕燕,罗红.葡萄球菌自溶素的研究进展[J].中国微生态学杂志,2011,23(3):272-274.
[4]You Yuan-hai.A novel DNA microarray for rapid diagnosis of enteropathogenic bacteria in stool specimens of patients w ith diarrhea[J].M icrobiol Methods,2008,75:566-571.
[5]Yoo SM,Lee S Y,Chang K H,et al.High-throughput identification of clinically important bacterial pathogens using DNA Microarray[J].Molecular Cellular Probes,2009,23(3-4):171-177.
[6]Chakravorty S,A ladegbam i B,Burday M,et al.Rapid universal identification of bacterial pathogens from clinical cultures by using a novel sloppy molecular beacon melting temperature signature technique[J].CliniM icrobiol,2010,48(1):258-267.
[7]Beck L F,Cardoso R.Development of a multiplex PCR assay for polymorphism analysis ofBrucella suisbiovars causing brucellosis in swine[J].Vet Microbiol,2006,115:269-277.
[8]Lu Zhi-tang,Wang Jing,Zhang Ya-ting.Quantitative real-time PCR detection of airborneStaphylococcus aureusin hospital indoor atmosphere[J].Modern App Sci 2012,6(3):22-26.
[9]Yauk C L.Comprehensive comparison of six Microarray technologies[J].Nucleic Acids Res,2004,32(15):e124.
[10]Ratushna V G,Weller JW,Gibas C J.Secondary structure in the target as a confounding factor in synthetic oligomer Microarray design[J].BMC Genom ics,2005,6(1):31.
[11]Rodaree K,Maturos T,Chaotheing S,et al.DNA hybridization enhancement using piezoelectric microagitation through a liquid coupling medium[J].Lab on a Chip,2011,6(11):1059-1064.
[12]Yoo SM,Lee S Y.Diagnosis of pathogens using DNA Microarray[J].Recent Patents Biotechnol,2008,2(2):124-129.
[13]Tian J,Ma K,Saaem I.Advancing high-throughput gene synthesis technology[J].Molecular Bio Systems,2009,7(5):714-722.
[14]Cai Ting,Zhang Shun,Li Qiao-yun,et al.Detection of common resistance genes of Gram-negative bacteria by DNA Microarray assay[J].African JM icrobiol Res,2012,6(2):371-378.
