浅谈新版GMP对建设洁净室(区)厂房的环境要求
2013-08-27康恺梁毅
康恺 梁毅
(中国药科大学国际医药商学院,江苏南京210009)
0 引言
我国新版GMP于2011年3月1日起开始正式执行。新版GMP对于“厂房与设施”的条例共有5节33条,分别是原则(第8条)、生产区(第11条)、仓储区(第6条)、质量控制区(第5条)和辅助区(第3条)。2010年的新版GMP与1998年版的GMP相比,在“厂房与设施”这一章从原来的23条增加到了33条,其中新增加条款12条,取消条款2条。
新版GMP在“厂房与设施”的条例中具体强调了厂房设计和布局的标准性和合理性,并具体从生产区、仓储区、质量控制区和辅助区这4个区域分别作不同的标准进行细化分类。其中,对洁净室的要求更加符合国际标准和我国的实际操作情况,有效地提高了药品生产过程中的硬件要求,保证药品生产过程中的质量。
1 新版GMP对厂房与设施的要求
新版GMP要求制药企业在生产过程中消除混药现象和污染现象,为此,制药企业必须具备与生产能力相适应的厂房与设施条件。这包括建立合格标准的洁净室(区)的空气处理系统、照明通风系统、卫生安全系统等,制药企业也采用洁净技术体系作为洁净室的重要组成部分以防止生产过程中的各种污染。而新版GMP要求所有制剂、原料药的精制、包装、干燥和所有直接接触药品的包装材料的生产等都必须在洁净室区域内完成。
1.1 生产过程中的洁净控制
制药企业的洁净室(区)的含义是指必须按照规定标准对微粒、微生物等污染进行严格控制的所有区域。药品有别于其他产品,药品生产的洁净室(区)必须包括以控制微粒污染为主要目的的工业洁净厂房要求和以控制微生物污染为主要目的的生物洁净室的要求。
微粒是指在药品生产过程中遇到的无生命的污染物,如尘粒等污染会影响药品质量,并在日后对人身安全造成危害。微生物是指在生产过程中遇到的有生命的污染物,如病毒、热原、细菌和过敏性物质等,它们产生和附着在厂房和设施上,会对药品造成污染,给环境造成危害。新版GMP要求下的洁净室(区)的微生物限制标准是严格按照中国药典(2010年版)中对药品微生物限制的要求进行实施,具体标准如表1所示。

表1 中国药典(2010年版)药品微生物限度标准表
1.2 生产环境的洁净控制
药品生产企业不仅要在生产过程中对洁净室(区)的污染进行控制,还要对生产环境进行污染控制。新版GMP将“无菌药品”生产洁净区分为以下4个级别[1]:
A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36~0.54m/s(指导值),应当有数据证明单向流的状态并经过验证。在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低的操作步骤的洁净区。
以上各个级别所对应洁净室(区)的洁净度级别如表2所示。
新版GMP对于高风险的“无菌药品”要求必须达到洁净室(区)悬浮粒子和表面微生物监测的动态标准和规定[2]。新版GMP第48条规定应当根据药品品种、生产操作要求及外部环境状况等配置空调净化系统,使生产区有效通风,并有温度、湿度控制和空气净化过滤,保证药品的生产环境符合要求。洁净区与非洁净区之间、不同级别洁净区之间的压差应当不低于10Pa。必要时,相同洁净度级别的不同功能区域(操作间)之间也应当保持适当的压差梯度。

表2 2010年新版GMP规定的洁净室洁净度级别
口服液体和固体制剂、腔道用药(含直肠用药)、表皮外用药品等非无菌制剂生产的暴露工序区域及其直接接触药品的包装材料最终处理的暴露工序区域,都应当参照“无菌药品”附录D级洁净区的要求设置,制药企业可根据产品的标准和特性对该区域采取适当的微生物监控措施,并按无菌药品、原料药、生物制品、血液制品、中药制剂5个附录分别提出洁净度要求,非无菌原料药精制、干燥、粉碎和包装等生产操作的暴露环境应当按照D级洁净区的要求设置;生物制品的洁净度要求见第10章无菌制剂生产设备的验证“生物制品生产操作实例”;血液制品的原料血浆破袋、合并、分离、提取、分装前的巴氏灭活等工序至少在D级洁净区内进行;中药制剂的浸膏的配料、粉碎、过筛、混合等操作,其洁净度级别应当与其配制操作区的洁净度级别一致;中药饮片经粉碎、过筛、混合后直接入药的厂房应当能够密闭,有良好的通风、除尘等设施,人员、物料进出及生产操作应当参照洁净区管理条例;中药注射剂浓配前的精制工序应当至少在D级洁净区内完成。
2 新版GMP建设洁净室(区)厂房的分析与设想
2.1 新版GMP洁净室(区)厂房要求达到国际化标准
新版GMP对洁净室(区)的空气洁净度要求不仅规范了我国制药企业的生产操作过程,同时也达到了WHO及其他发达国家的GMP要求,具体的国内外标准如表3所示。GMP实施的终极目的是将生产操作过程中的人为差错减小到最低程度,以有效地防止药品受到污染,从而实现健全的质量保证体系[3]。在新版GMP实施过程中,必须站在推行多年的旧版GMP基础之上,不能脱离了中国的实际发展水平,同时又要迅速与国际接轨,这对于我国制药企业来说是一个全新的挑战。
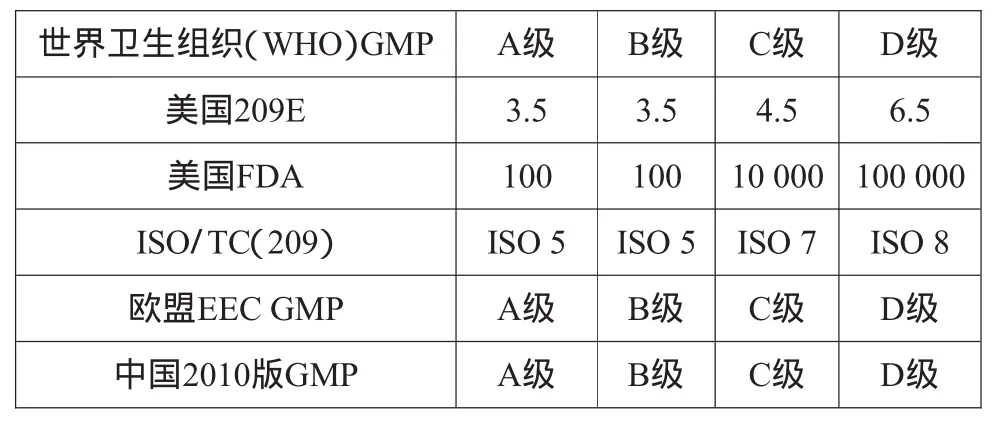
表3 中国2010版GMP洁净度实施标准级别与国外实施标准对比
2.2 无菌药品的洁净室(区)构建难度颇大
新版GMP对于高风险的“无菌药品”要求必须达到洁净室(区)悬浮粒子和表面微生物监测的动态标准和规定。所以新建厂房洁净室(区)应该按照最高级(A级)标准建造。但是,我国的实际水平和旧版GMP的多年实行情况,使得我国制药企业在洁净室(区)的设计、安装、运行和管理方面都缺乏先进经验,很难达到新版GMP的要求。例如我国制药企业习惯于按照顶送侧回的送回风方式来建造洁净室(区),但当厂房换气次数有所提高,顶送底回即地面格栅回风的方式就不能满足洁净室(区)的换气要求。这样一来,如果对厂房进行改造,不仅会使洁净室(区)构建结构改变,在厂房管理上也会产生一系列新的问题。
2.3 控制洁净室(区)的最大污染源
操作人员是药品生产过程中最大的污染源也是最主要的传播媒介,人员直接或间接地接触药品都会对药品的质量产生严重影响,洁净室内微粒来源如表4所示,其影响因素包括2个方面,一个方面是人员身体状况所产生的污染;另一个方面是人员的个人卫生习惯所造成的污染。所以,控制好人员的卫生管理和监督是有效保证药品质量的关键因素。
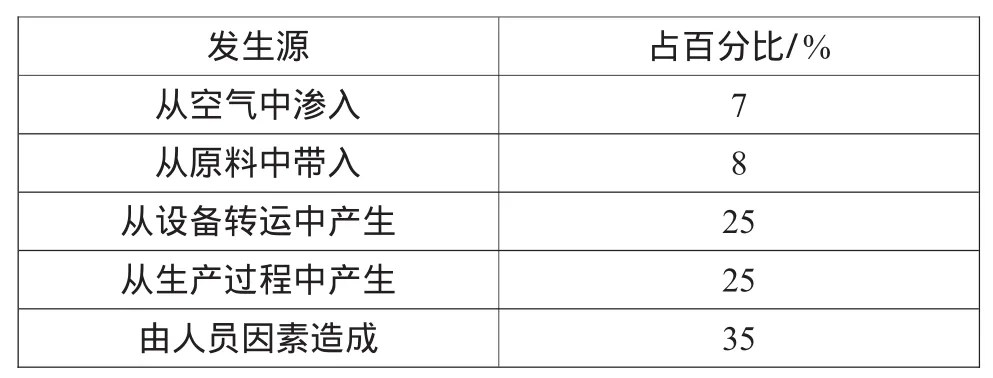
表4 洁净室内微粒来源分析表
新版GMP第3章第4节“人员卫生”共计9条标准,严格要求和控制了人员卫生造成的厂房洁净室(区)的污染,其规定了人员必须按照“良好卫生规程(GoodHygiene Practice,GHP)的8项基本操作法进行操作,还必须建立个人卫生档案。在欧美发达国家的GMP章程中还规定从事高敏感区域的生产人员必须每6个月做1次blood test(血常规检查);从事视觉检查岗位的人员必须进行eyetest(视力测试)等。
在防止人员产生污染的同时,减少操作人员人数及降低人员活动频度是十分重要的。对于可不要求人员参与的生产工序(如灌装、加塞等制作工序),可以依靠高性能的设备单独完成,以避免造成人员污染。同时严格维持相邻不同等级洁净区之间的压差不得低于10Pa、洁净区与非洁净区的压差不得低于15Pa,以此减少尘埃粒子的渗入,尽可能减少尘埃粒子的产生是以最少换气次数达到高等级洁净度最经济的方法。
3 结语
洁净室(区)的厂房建设是新版GMP的主要硬件部分,硬件是GMP实施的基础,而硬件是否符合标准直接影响药品的生产质量[4]。我国制药企业在建设洁净室(区)的厂房时不仅要严格参照新版GMP操作规范来进行操作,同时还要按照GB50457—2008《医药工业洁净厂房设计规范》来科学建设厂房,只有在设计质量和施工质量都达到洁净室(区)厂房中符合操作标准的制作工艺,才能使得药品质量有所保障,从而推动企业实现利益最大化。
[1]李钧,李志宁.制药质量体系及GMP的实施[M].化学工业出版社,2011
[2]梁静频,王燕祖.GMP修订带来洁净厂房的变革[J].中国医药生物技术,2009,2(4)
[3]梁毅.制药企业实施GMP认证及其意义[J].机电信息,2012(20)
[4]梁毅.新版GMP教程[M].北京:中国医药科技出版社,2011
