单分散掺硫碳微球的水热制备及其表征
2013-08-20郑明涛张浩然董汉武龚学斌徐汝淳雷炳富刘应亮刘晓瑭
郑明涛 肖 勇 张浩然 董汉武 龚学斌 徐汝淳 雷炳富 刘应亮 刘晓瑭
(华南农业大学理学院应用化学系,广州 510642)
0 引 言
碳微球(Carbon microspheres, CMSs)由于其高比表面积、高堆积密度、化学惰性、易石墨化片状炭层结构的球体、自烧结性能良好等优良性能,可广泛应用于催化剂载体、锂离子电池、电化学电容器、抗菌材料载体、化学模板、高密高强碳材料等领域,是一种具有广泛应用前景的碳材料,成为当前碳材料研究的热点之一[1-10]。目前,科研人员采用化学气相沉积[11]、高温碳化[12]、催化热解[13]、金属碳化物还原[14]、水热碳化[15]、溶剂热反应[16]等方法成功地制备了不同粒径大小的CMSs。其中,水热碳化法采用廉价的碳水化合物为原料以及所制备的材料表面含有丰富的含氧基团等特点而成为一种新兴的碳功能材料制备方法,采用该方法制备CMSs 也已经得到了一定的研究[17-25],但是关于制备均匀性高、分散性 好 的 掺 硫CMSs(sulfur-doped CMSs,SCMSs)的 研究却鲜见文献报道。本研究通过在水热碳化体系中引入硫单质,成功地开辟了一条单分散SCMSs 的合成路线,其操作简单、重复性好。更重要的是,采用硫辅助水热碳化法能极大地提高SCMSs 的产量,同时可以同步将硫原子掺入碳微球的网络结构中。本文重点分析讨论了硫掺杂对产物微观结构的影响,并对硫辅助水热碳化条件下SCMSs 的形成过程进行了简要分析。
1 实验部分
1.1 原料及仪器
实验所用的原料可溶性淀粉、升华硫、无水乙醇均为国产分析纯试剂,溶剂选用实验室自制的去离子水,采用不锈钢水热反应釜作为反应发生器(最高耐温:700 ℃,最高耐压:40 MPa)。
1.2 制备过程
SCMSs 的制备: 将20 g 可溶性淀粉,4 g 升华硫,20 mL 去离子水调成匀浆;再将匀浆倒入60 mL容积的不锈钢反应釜,密封好的反应釜置于程序控温井式炉中以10 ℃·min-1的升温速率加热到550℃,并在550 ℃恒温6 h,然后自然冷却至室温。CMSs 的制备与文献[24]报道的实验过程相似:将4 g淀粉,40 mL 去离子水加入到60 mL 容积的不锈钢反应釜。反应温度、升温速率和恒温时间与SCMSs相同。将反应釜中黑色的物质取出,经去离子水、乙醇洗涤,离心分离,然后在60 ℃真空干燥8 h。最后所得的产物经称量后,其质量分别为6.2 g (SCMSs)和0.9 g (CMSs)。
1.3 产物的表征
采用荷兰PHILIPS XL-30S 扫描电子显微镜(SEM,加速电压30 kV)和日本电子JEM-2010 型透射电子显微镜(TEM,加速电压200 kV,Cu 网,碳膜)观察产物的微观形貌。通过MSAL-XD2 全自动粉末X 射线衍射仪表征产物的物相结构,采用Cu Kα 射线(λ=0.154 06 nm)。采用INCA X200 型能谱分析仪(EDS)、ESCALAB 250 型X 射线光电子能谱仪(XPS)分析产物的元素组分。采用显微激光Raman 光谱仪(Renishaw,514.5 nm 氩离子激光器) 测试材料的Ranman 光谱。采用傅里叶变换红外光谱(FTIR,Thermo Electron Corporation)测试产物表面残留的有机基团。粉末的核磁共振13C 谱在ANCE AV 400M NMR(Bruker)核磁共振仪上测得。采用美国Tristar 3000 全自动比表面和孔隙分析仪测定材料的比表面积和孔径分布。采用Q600 SDT(TA Company)热重分析仪测试样品的热稳定性。
2 结果与讨论
2.1 产物的形貌及EDS 分析
产物的形貌和微观结构采用SEM 和TEM 电镜来进行分析表征。从图1a 中的低倍SEM 照片可以看出,样品中SCMSs 的纯度高,碳球含量接近100%。图1b 是高倍SEM 照片,可以看出,通过这种方法得到的SCMSs,分散性良好,表面光滑,未见其它形貌碳材料存在,说明此法是制备高质量、高纯度SCMSs 的有效途径。SCMSs 的粒径分布均匀,约为4.0 μm。图1c 是SCMSs 的TEM 图片。从图可以看出,所得SCMSs 由粒径约为4.0 μm 的实心球组成,粒径分布均匀,与SEM 观察结果一致。图1d 是SCMSs 球壁的高分辨TEM 图片。从图可以看出,SCMSs 并不具有良好的晶态石墨层结构,而是无定形结构。图1d 中的插图是相应的选区电子衍射花样,弥散的衍射环花样表明产物是无定形的。图1e和f 是采用水热碳化法所制备的CMSs 的SEM 图,可以看出,所得产物为粒径约为2.0 μm 的球形颗粒,这与文献[24]报道的一致。研究结果表明,在水热碳化体系中引入硫,能有效地提高单分散SCMSs 的产量。
从图2 中的EDS 图谱可以看出,所制备的SCMSs 和CMSs 中主要含有C 和O 两种元素。采用硫辅助水热碳化法制备的SCMSs 含有约2.00At%的硫元素,其中少量的Fe 可能来源于硫化氢与不锈钢反应釜反应生成的FeS。而在CMSs 中除了含有C 和O 外不含有其它元素。EDS 研究结果表明,这种硫辅助水热碳化法能在合成过程中同步将硫原子掺入碳微球的网络结构中。

图1 硫辅助水热碳化法制备的SCMSs (a, b)不同标尺的扫描电镜图,(c) 透射电镜和(d)高分辨透射电镜图,(e, f) 传统水热碳化法制备的CMSs 的扫描电镜图,图d 中插图是SCMSs 相应的选区电子衍射花样Fig.1 (a, b) SEM, (c) TEM, and (e) HRTEM images of the sulfur-doped carbon microspheres (SCMSs)by sulfur-assisted hydrothermal carbonization (SAHTC), (e, f) SEM image of the as-prepared carbon microspheres (CMSs) by hydrothermal carbonization (HTC). Inset in d shows the corresponding selected area electronic diffraction pattern of SCMs
2.2 XRD 与FTIR 分析
图3 为所得SCMSs(a)和CMSs(b)的XRD 图。图中2θ=25.7 和43.1°处存在的2 个衍射峰分别对应石墨的(002)和(101)衍射(PDF No.75-1621)。与CMSs相比,SCMSs 的(002)衍射峰的位置向低角度偏移,说明石墨层片间距增大,平均晶粒尺寸减小。从图可以看出,(002)晶面的衍射峰强度较低,表明制备的SCMSs 石墨化程度不高,属于无定形态结构。这主要是由以下原因造成的:(1) 合成温度相对较低;(2) 碳来自于淀粉水解产物的脱水碳化,在没有金属催化剂的情况下裂解制备的碳材料比较难以石墨化。
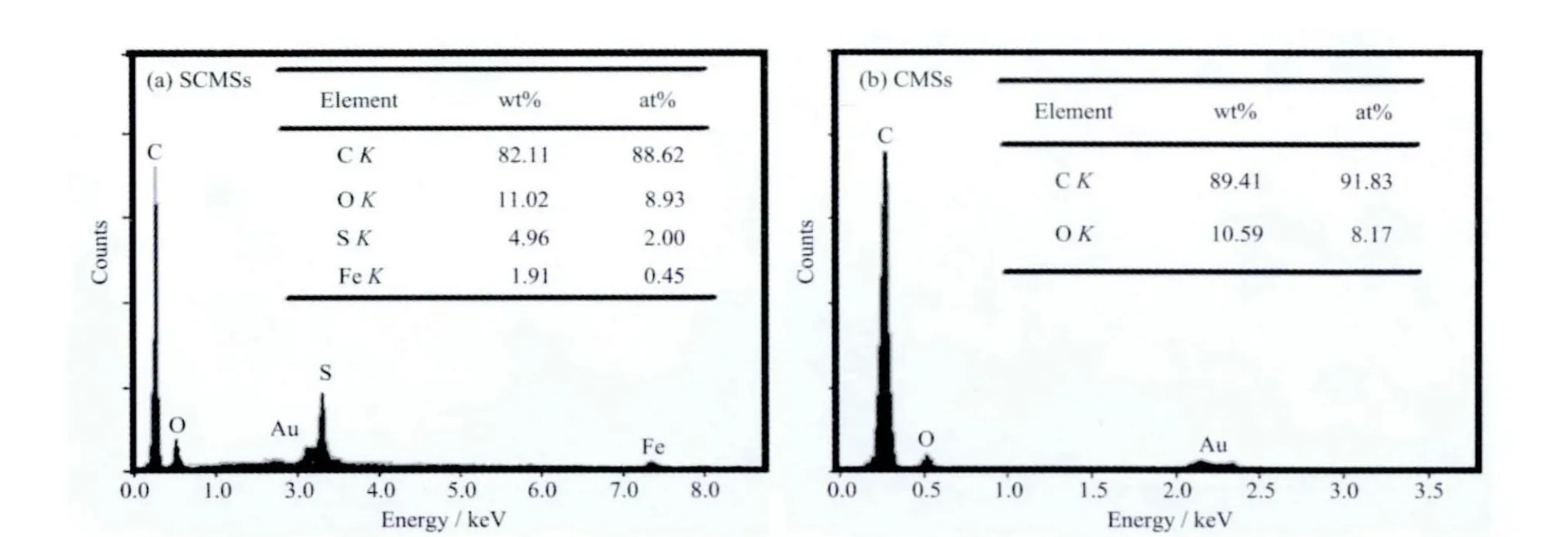
图2 不同样品的EDS 图谱Fig.3 EDS spectra of the as-prepared samples
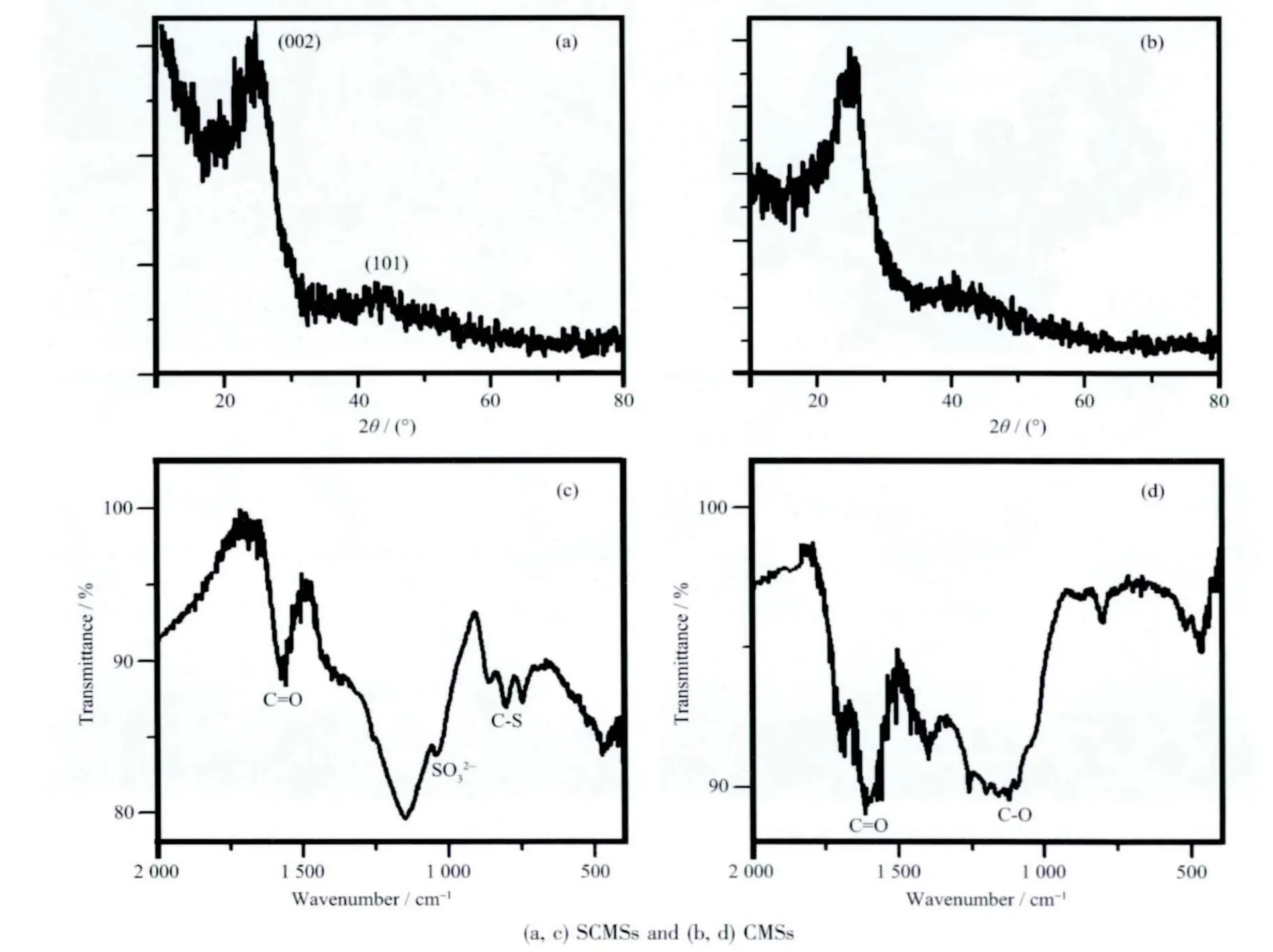
图3 SCMSs 和CMSs 的(a, b)X 射线衍射和(c, d)红外光谱Fig.3 (a, b) XRD patterns and (c, d) FTIR spectra of the as-prepared samples
产物的傅里叶变换红外光谱如图3c 和d 所示。可以看出,SCMSs 和CMSs 的表面均含有丰富的有机官能团。在FTIR 图谱中,位于1 650 cm-1附近的吸收峰可以归属为C=O 振动,而1 150 cm-1附近的吸收峰属于C-O 的振动吸收。一般来说,C=O 的吸收峰要强于C-O 基团,但是在SCMSs 中,位于1 180 cm-1附近的吸收峰却要强于1650 cm-1峰,其可能的原因在于O=S=O(亚磺酸基或磺酸基)在此范围内存在较强的振动吸收,磺酸基是双峰吸收,它的孪峰位于1 043 cm-1附近。位于900~700 cm-1范围的3 个吸收峰(855、809、784 cm-1) 可归属于C-S的振动吸收。S=O 和C-S 的存在表明硫在SCMSs 中与碳形成化学键,同时也说明采用硫辅助水热碳化法可以成功将硫原子掺入碳球的网络结构中。
2.3 显微Raman 分析
激光显微Raman 光谱法是一种无损检测碳材料缺陷和无序性的常用方法。通过确认是否有D 峰来确定缺陷和无序性。因为在单晶石墨中只有1 个峰G 峰,而无序的石墨有2 个峰: 在1 580~1 600 cm-1的G 峰和1 350 cm-1附近的D 峰[26-28]。如图4所示,SCMSs 和CMSs 都含有明显的G 峰和D 峰,在1 597 cm-1的G 峰是E2g对称中心区域振动,在1 200~1 400 cm-1是A1gK-point 的声子振动。G 峰与共面sp2杂化碳对所成键的振动有关,它发生在sp2杂化的芳香或者烯烃的分子中[29]。如图4a 所示,SCMSs 的D 峰可以拟合为3 个峰: 分别位于1 289 cm-1(D1),1 360 cm-1(D2)以及1 430 cm-1。其中,D1是典型的由于弧形结构造成芳香基团的边缘振动[29-30],D2峰与引入缺陷的数量有关,也是与无序性相关的峰。而位于1 430 cm-1附近的拉曼峰,此峰在carbon peapods[31]中认为是“pentagonal pinch”五边形挤压,类似的五边形结构在掺硫碳纳米管中也可以观察到[32-33]。如图4b 所示,CMSs 的D 峰可拟合为2个峰:1 289 cm-1(D1)和1 360 cm-1(D2)。这与SCMSs是不同的,表明在CMSs 中很少有类似的五边形结构存在。Tuinstra 和Koenig[26]提出D 峰与G 峰的强度之比ID/IG与石墨的晶粒尺寸La成反比。在本研究中ID/IG(SCMSs)=1.35,ID/IG(CMSs)=1.15,表明SCMSs 的晶粒尺寸比CMSs 的略小。晶粒尺寸由六元环减小到五元环,这与观察到的“pentagonal pinch”五元环的1430 cm-1附近的拉曼吸收峰吻合,表明硫原子掺杂对SCMSs 的微观结构产生了较大的影响。
2.4 XPS 分析
产物的表面元素组成和状态采用XPS 进行分析,如图5 所示。在XPS 图谱中,SCMSs 含有碳、氧以及硫3 种元素,而CMSs 只含有碳和氧2 种元素。C1s 峰位于284.6 和285.5 eV 分别认为是sp2和sp3杂化的碳。位于289.3 eV 峰认为是C=O(酮,内酯,羰基)[34-35]。位于286 eV 的峰在CMSs 中可以归属为C-O,而在SCMSs 中则归属为C-S,因为C-O(534.10 eV)的能级较C=O(532.73 eV)的要高,而SCMSs 在此位置(534.10 eV)没有峰存在。C-O 峰可能来源于微球表面吸附的醚和醇,这与FTIR 分析结果相吻合。在S2p 图谱中(图5c),162.46 eV (18.4At%)、163.06 eV(4.9At%)、163.71 eV(27.18At%)、164.31 eV(20.20At%)、167.76 eV(29.29At%)分 别 对 应 于PhSR和PhSH、PhSPh、PhSSPh、噻吩、SO2-和SO22-[34-35]。以上 研 究 结 果 表 明:PhSSPh、 噻 吩、SO2-和SO22-是SCMSs 中硫掺杂的主要形式。
2.5 13C 核磁共振分析

图4 所制备样品的Raman 光谱(a) SCMSs, (b) CMSsFig.4 Raman spectra of the as-prepared (a) SCMSs and (b) CMSs
所制备的SCMSs 和CMSs 的C13环境采用CP MAS NMR 进行表征。如图6 所示,特征峰127 显示CMSs 是sp2杂 化 的 石 墨 化 的 内 核[36]。SCMSs 和CMSs 都在188 含有C=O。位于127、150、188 的3个峰证明了杂原子的存在,但是C-O 与C-S 的化学位移很难区分,与之相似的有苯甲酸(δCOOH=173)、苯酚(δC-OH=155)[37]、苯 磺 酸(δC-SO3H=146)、苯 硫 醇(δC-SH=131)、噻 吩(δC-S=126)、1,2-diphenyldisulfane(δC-S=136)。约19 ppm 是脂肪烃末端甲基峰。一般认为脂肪烃中CH3(5~20)的峰强小于或等于CH2(~40)的峰强,如在CMSs 中CH3与CH2的峰强近乎相等。但是在SCMSs 中,CH3的峰强明显大于CH2,其可能的原因是当硫原子取代六元环上的碳原子时,由于边缘六元环的内聚力,部分C-S 键断裂重新成键,生成五元噻吩环和甲基。硫原子在SCMSs 中的主要存在形式如图7 所示,从硫的角度来看,插入到片层间的硫基团如:-S-S-、-S-、-SO2-和-SO-对于小的石墨烯片起到联结剂的作用,而从完美六元环石墨的角度看含硫基团构成杂原子缺陷。
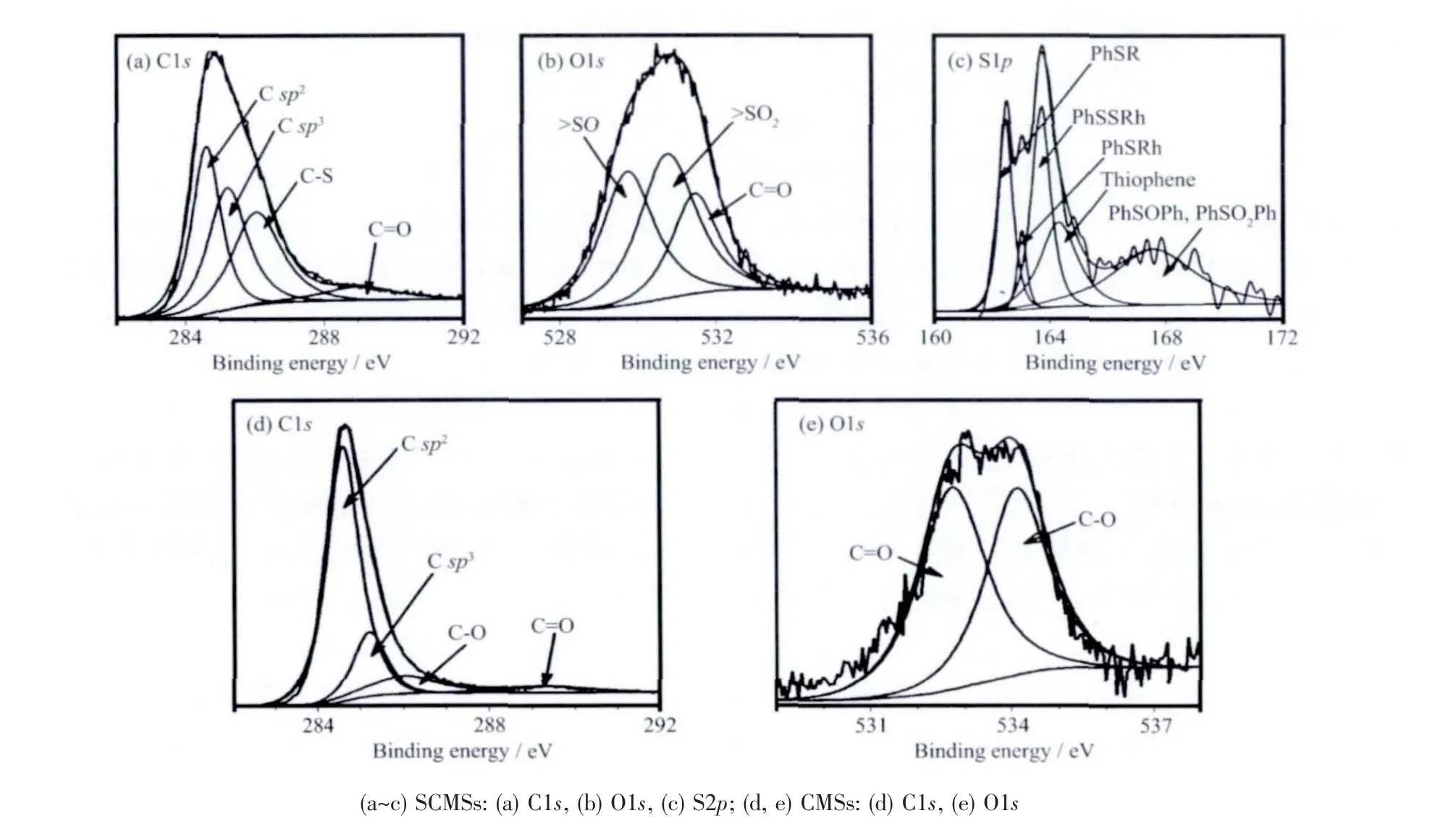
图5 所得产物的X 射线光电子能谱Fig. 5 XPS spectra of the as-prepared SCMSs and CMSs
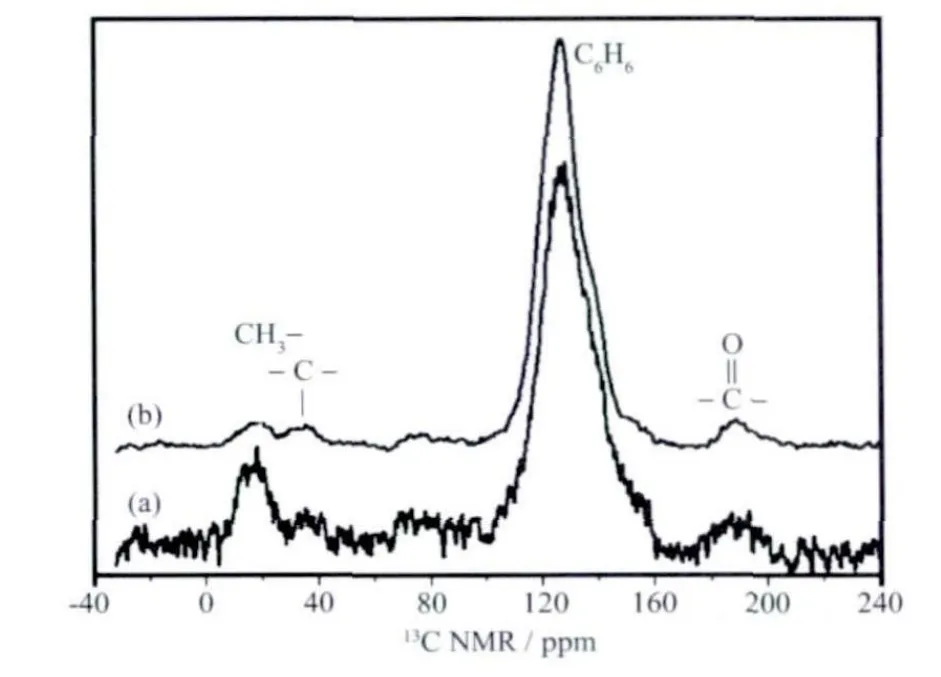
图6 (a) SCMSs 和(b) CMSs 的固体13C NMR 谱Fig.6 13C NMR spectra of the as-prepared (a) SCMSs and (b) CMSs
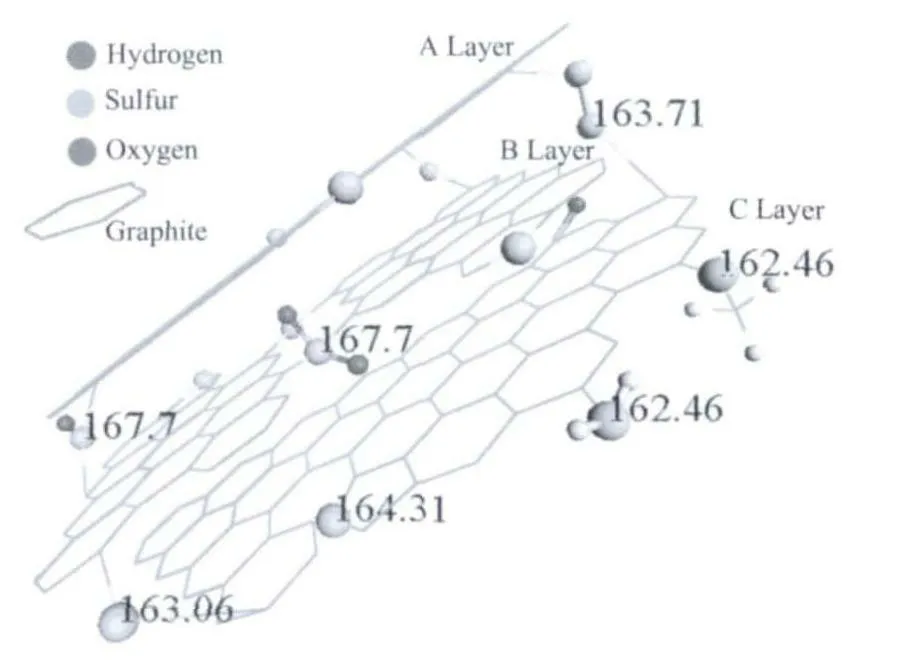
图7 硫在SCMSs 中的存在形式示意图Fig.7 Schematic illustration of the existing forms of sulfur in SCMSs
2.6 比表面及孔径分析
所得样品的N2吸脱附曲线及孔径分布如图8所示。SCMSs 和CMSs 的BET 比表面积分别为443和23.6 m2·g-1,平均孔径分别为3.5 和2.3 nm。从图8b 可以看出,SCMSs 的孔径除了在1.8 nm 分布外,在4.2 nm 也有明显的分布,而CMSs 的孔径分布则主要集中在2.3 nm 左右。分析结果表明,采用硫辅助水热碳化法所制备的SCMSs 的比表面积是采用普通水热碳化法制备的CMSs 的近20 倍。这可能由于硫原子的掺杂效应: 噻吩、-S-S-、-S-、-SO2-和-SO-等增加了SCMSs 对气体的吸附点,同时-S-S-、-S-、-SO2-和-SO-等插入到石墨烯片层之间扩大了SCMSs 中石墨层的面间空间,从而增大了SCMSs 的比表面积。由于SCMSs 具有高的表面积,这种新型碳材料在气体存储、催化剂载体以及能量存储等领域中将具有广泛的应用前景。
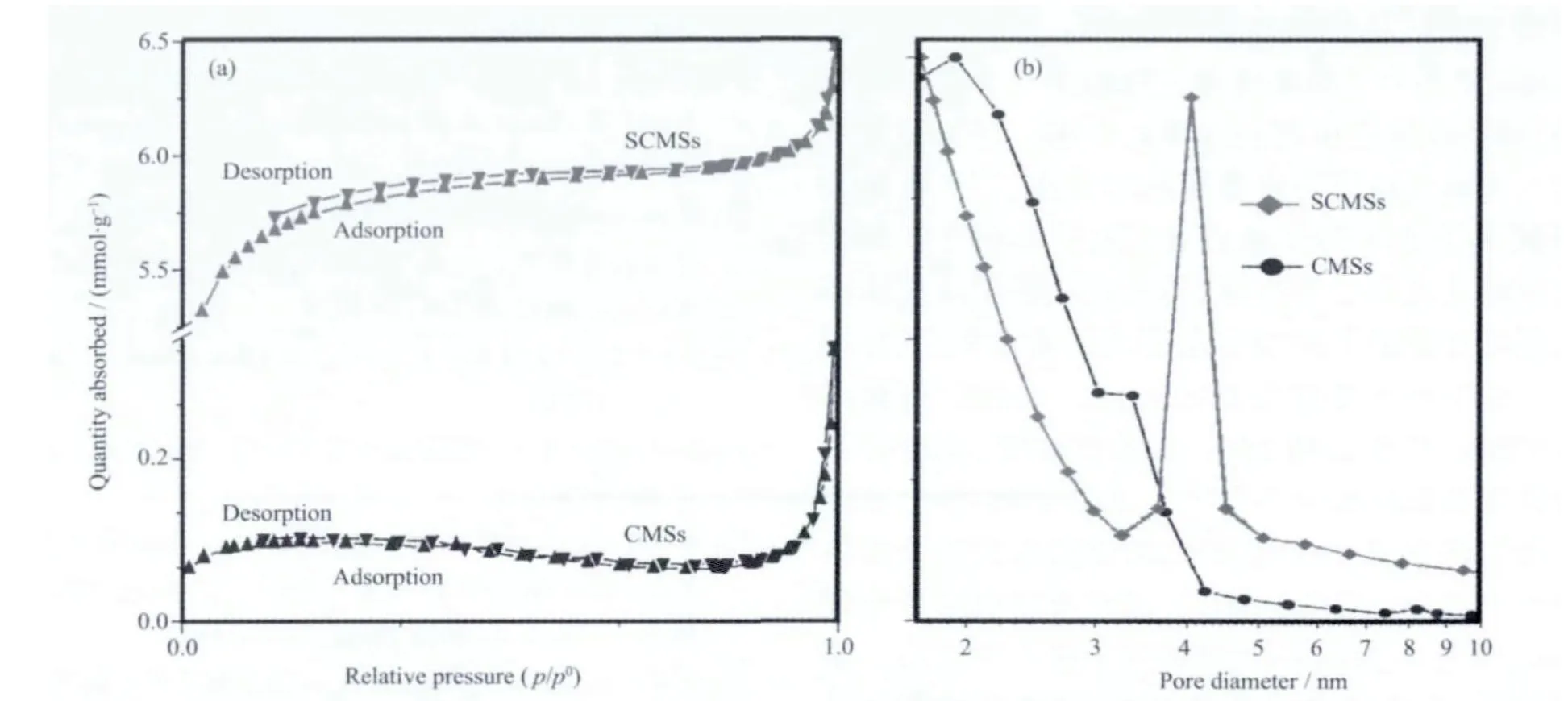
图8 所制备的SCMSs 和CMSs 的N2 吸脱附曲线(a)和孔径分布图(b)Fig.8 N2 absorption/desorption isotherms (a) and pore size distribution (b) of the as-preparedSCMS and CMSs
2.7 热稳定性测试
图9 是所制备的SCMSs 和CMSs 球的热重和微商热重曲线。从图9a 可以看出,SCMSs 的热分解可以分为300~375、380~473、480~743、750~1200 K四阶段。第一步为SCMSs 表面吸附的水和硫醇的脱附;第四步与CMSs 相似,是碳球表面含氧基团的分解。显然,第二步和第三步与掺杂的硫有关,我们认为第二步为S-S 的断裂,第三步为C-S 键的断裂。这进一步说明了在SCMSs 中噻吩、-S-S-、-S-、-SO2-和-SO-等基团的存在。从图9b 可以看出,CMSs 对热稳定直到750 K,从750~1 100 K 随着温度的升高质量减少,整个过程质量减少约30wt%。对SCMSs和CMSs 的热稳定性分析结果表明: 相对CMSs 而言,SCMSs 对热相对不稳定。
2.8 SCMSs 的形成过程

图9 (a) SCMSs 和(b) CMSs 的热重和微商热重(TG-DTG)曲线Fig.9 TG-DTG curves of the as-prepared SCMSs (a) and CMSs (b)
水硫热反应(hydro-sulfur reaction)在地质化学、工程技术、环境科学等领域研究广泛[38-42]。在823 K下,与传统的超临界水热反应,水硫热反应更加活泼。在本研究中,可溶性淀粉和升华硫在水溶液里加热到823 K,达到超临界状态[41-42]。超临界流体由H2O-H2S 和小分子的硫组成。这些小分子硫由升华硫热分解,在反应中起到重要的作用。首先小分子硫参与水解反应以及自氧化还原反应,产生硫化氢和磺酸[39];其次小分子硫还会从有机物中夺取氢原子生成硫化氢和二氧化碳;另外硫交联反应连接不同石墨烯片促进了SCMSs 的形成。通常情况,在水热反应中淀粉的浓度为0.10 g·mL-1时可以得到单分散CMSs,产率为约25%,超过此浓度,碳球容易团聚结块而且此反应不可逆[24]。而在本研究中,在硫存在的条件下,反应物淀粉的浓度提高到传统水热碳化水热的10 倍(10 g·mL-1),同样可以得到单分散SCMSs,可见SCMSs 的产量得到了极大的提高。更重要的是,在水热碳化的过程中,硫原子能有效地掺入碳球的网络结构中,对碳球的微观结构产生重要的影响: 噻吩、-S-S-、-S-、-SO2-和-SO-等基团构成了SCMSs 的化学缺陷,而噻吩等五元环取代六元石墨环构成了SCMSs 的结构缺陷,这些缺陷的形成极大地增大了SCMSs 的比表面积,同时也将对其性能产生重要的影响。
3 结 论
本文以可溶性淀粉为碳源,升华硫为硫源,采用水热碳化法一步合成出了平均直径约为4 μm 的单分散SCMSs。研究结果表明,在硫单质存在的条件下,采用水热碳化法可以高浓度、高产量地制备单分散SCMSs,更重要的是硫原子可以同步掺入碳球的网络结构中。相对传统水热碳化法而言,采用硫辅助水热碳化法制备的SCMSs 具有更高的比表面积,其可能的原因是硫原子的掺杂产生了大量的化学和结构缺陷。研究认为,-S-S-、-S-、-SO2-和-SO-等基团构成了化学缺陷,而噻吩等五元环取代石墨六元环构成了结构缺陷。含硫基团联结不同石墨烯片使SCMSs 容易形成,而填充在石墨烯片间的含硫基团扩大了石墨的面间距,在石墨烯片上五元的噻吩结构取代六元的石墨单元,减小了晶粒尺寸。
[1] CHENG Li-Qiang(程立强),LIU Ying-Liang(刘应亮),ZHANG Jing-Xian(张 静 娴), et al. Prog. Chem.(Huaxue Jinzhan),2006,18(10):1298-1304
[2] Xu C W,Cheng L Q,Shen P K,et al. Electrochem.Commun.,2007,9:997-1001
[3] WANG Xiu-Li(王秀丽), LIN Hong-Yan(林宏艳), LU Hai-Yan(卢海燕),et al.Chin.J.Appl.Chem.(Yingyong Huaxue),2006,23(11):1223-1227
[4] PING Li-Na(平丽娜), ZHENG Jia-Ming(郑嘉明), SHI Zhi-Qiang(时 志 强), et al. Acta Phys.-Chim. Sin.(Wuli Huaxue Xuebao), 2012,28(7):1733-1738
[5] Pan L L, Bi J Q, Bai Y J, et al. J. Phys. Chem. C, 2008,112:12134-12137
[6] Deshmukh A A, Mhlanga S D, Coville N J. Mater. Sci. Eng.R, 2010,70:1-28
[7] WU Yong-Jian(武拥建), ZHENG Ming-Tao(郑明涛), XIE Chun-Lin(谢 春 林), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2011,27(12):2447-2452
[8] CHEN Feng(陈丰), CHEN Zhi-Gang(陈志刚), LI Xia-Zhang(李 霞 章), et al. J. Chin. Ceram. Soc.(Guisuanyan Xuebao),2011,39(3):397-402
[9] CI Ying(慈颖), GE Jun(葛军), Wang Xiao-Feng(王小峰),et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2007,23(2):365-368
[10]Cui J, Hu C, Yang, et al. J. Mater. Chem., 2012,22:8121-8126
[11]Serp P, Feurer R, Kalck P, et al. Carbon, 2001,39:621-626
[12]Zhao S, Wang C Y, Chen M M, et al. Mater. Lett., 2008,62:3322-3324
[13]Wang Z L, Kang Z C. J. Phys. Chem., 1996,100:17725-17731
[14]Lou Z S, Chen Q W, Gao J, et al. Carbon, 2004,42:229-232
[15]Wang Q, Li H, Chen L Q, et al. Carbon, 2001,39:2211-2214
[16]YIN Cai-Liu(尹彩流), HUANG Qi-Zhong(黄启忠), WANG Xiu-Fei(王 秀 飞), et al. Carbon Techn.(Tansu Jishu), 2009,28(3):19-22
[17]Sun X M, Li Y D. Angew. Chem. Int. Ed., 2004,43:3827-3831
[18]Sun X M, Li Y D. Angew. Chem. Int. Ed., 2004,43:597-601
[19]Cui X J, Antonietti M, Yu S H. Small, 2006,2:756-759
[20]Sevilla M, Fuertes A B. Carbon, 2009,47:228-229
[21]Sevilla M, Fuertes A B. Chem. Eur. J., 2009,15:4195-4203
[22]Baccile N, Laurent G, Babonneau F, et al. J. Phys. Chem.C, 2009,113:9644-9654
[23]Mi Y Z, Hu W B, Dan Y M, et al. Mater. Lett., 2008,62:1194-1196
[24]Zheng M, Liu Y, Xiao Y, et al. J. Phys. Chem. C, 2009,113:8455-8459
[25]Zheng M, Liu Y, Jiang K, et al. Carbon, 2010,48:1224-1233
[26]Tuinatra F, Koenig J L. J. Chem. Phys., 1970,53:1126-1130
[27]Ferrari A C, Robertson J. Phys. Rev. B, 2000,61:14095-14107
[28]Ferrari A C. Solid State Commun., 2007,143:47-57
[29]Compagnini G, Puglisi O, Foti G. Carbon, 1997,35:1793-1797
[30]Tan P, Dimovski S, Gogotsi Y. Phys. Eng. Sci., 2004,362:2289-2310
[31]Chadli H, Rahmani A, Sbai K, et al. Phys. A, 2005,358:226-236
[32]Lu X, Sun C, Li F, et al. Chem. Phys. Lett., 2008,454:305-309
[33]Tapasztó L, Kertész K, Vértesy Z, et al. Carbon, 2005,43:970-977
[34]Yue Z R, Jiang W, Wang L, et al. Carbon, 1999,37:1785-1796
[35]Puziy A M, Poddubnaya O I, Socha R P, et al. Carbon,2008,46:2113-2123
[36]Cai W, Piner R D, Stadermann F J, et al. Science, 2008,321:1815-1817
[37]Hayashia S, Hoshib F, Ishikurab T, et al. Carbon, 2003,41:3047-3056
[39]Bondarenko G V,Gorbaty Y E. Geochimica et Cosmochimica Acta, 1997,61:1413-1420
[40]Tsuchiya N, Suto Y, Kabuta T, et al. J. Mater. Sci., 2008,43:2115-2122
[41]Gorbaty Y E, Gupta R B. Ind. Eng. Chem. Res., 1998,37:3026-3035
[42]Gorbaty Y E, Kalinichev A G. J. Phys. Chem., 1995,99:5336-5340
