两种组成型启动子P ermE*和P Sau3A在变铅青链霉菌中的启动效果比较
2013-08-16陶欣艺王风清魏东芝
张 莹,陶欣艺,王风清,魏东芝
(华东理工大学生物反应器工程国家重点实验室鲁华生物技术研究所,上海200237)
两种组成型启动子P ermE*和P Sau3A在变铅青链霉菌中的启动效果比较
张 莹,陶欣艺,王风清,魏东芝
(华东理工大学生物反应器工程国家重点实验室鲁华生物技术研究所,上海200237)
红霉素抗性启动子P erm E*和来源于链霉菌核基因组片段的P Sau3A启动子均属于链霉菌组成型强启动子,分别将它们插入大肠杆菌-链霉菌穿梭质粒p NW-S1的SD序列和转录起始位点之间,构建成两个稳定的组成型表达质粒:p NW-S3和p NW-S4。以磷脂酰丝氨酸合成酶PSS基因为报道基因,以蛋白表达水平评价这两个启动子在变铅青链霉菌TK24中的表达效果。结果表明,在两株组成型表达重组菌中,PSS均得到了高效表达,具有在链霉菌系统中高效表达外源基因的功能,其中P erm E*的启动效率略高于P Sau3A。
组成型启动子;链霉菌;磷脂酰丝氨酸合成酶
链霉菌属于放线菌的一科,是G+C含量较高的革兰氏阳性菌,可生产多种抗生素,同时还能分泌大量的胞外蛋白,如各种水解酶和蛋白类的酶抑制剂等,广泛应用于工业生产中,特别是食品医药行业。链霉菌的高效分泌能力、人畜的非致病性以及成熟的发酵工艺使其成为表达异源蛋白的新宿主[1]。
作为一种具有巨大应用价值的生产菌,链霉菌的代谢过程是一个复杂的、互相偶联的多层次调控过程,而启动子和其它顺式调控序列构成了这个调控系统中的基础部分[2]。链霉菌工程菌的启动子多为诱导型启动子,其表达效果虽然优于一般的组成型启动子,但是需要IPTG、硫链丝菌素等诱导剂的诱导,增加了链霉菌作为安全生产宿主的局限性,对于表达一些有安全要求的目的基因时,增加了后期分离纯化和医药审批的难度,限制了其工业化应用。因此,构建高效安全的组成型链霉菌表达载体,用以表达医药食品行业中有安全要求的基因,具有重要的实际意义。
目前,链霉菌常用的组成型启动子是红霉素抗性启动子P erm E,尤其是改造过的P erm E*启动子[3,4],而对其它组成型启动子的研究较少。Horinouchi等[5]筛选到一个来源于链霉菌的约120 bp的基因片段[6],具有较高的启动子活性,是红霉素启动子P erm C活性的2.4倍。作者选用常用的链霉菌异源基因表达宿主变铅青链霉菌(Streptomyces lividans) TK24作为转化受体,P erm E*和P Sau3A作为启动子,以磷脂酰丝氨酸合成酶(PSS)作为报告基因,分别构建两种不同的组成型表达载体,评价比较这两种启动子的启动效率,以期构建高效安全的链霉菌组成型表达质粒,应用于工业生产。
1 实验
1.1 材料
1.1.1 菌株及质粒
大肠杆菌DH5α购于天根生化科技公司。含有红霉素抗性启动子P ermE*的大肠杆菌-链霉菌穿梭质粒pIJ4090和表达菌株变铅青链霉菌TK24为自行保存。大肠杆菌-链霉菌穿梭表达质粒pNW-S1为前期构建。
1.1.2 链霉菌启动子
红霉素抗性强启动子Perm E*来源于pIJ4090质粒。在寻找效果更好的组成型启动子时,发现来源于Streptomyces griseus的具有强启动子活性的基因片段,约1.6 kb,在保证后续研究中启动子活性不降低的情况下,最终确定该片段长度为124 bp。将该段序列命名为P Sau3A,由捷瑞生物公司进行基因合成,以连接在p GH质粒(p GH-P Sau3A)上的形式返回。
1.1.3 磷脂酰丝氨酸合成酶基因
链霉菌来源的磷脂酰丝氨酸合成酶PSS基因为前期克隆保存。
1.1.4 培养基
LB培养基用于大肠杆菌的培养和重组质粒的提取。TBS培养基用于链霉菌的发酵培养。R2YE培养基用于链霉菌的固体斜面培养[7]。CP培养基用于链霉菌原生质体的制备[8]。
1.1.5 工具酶、PCR酶以及引物
限制性内切酶购于Takara公司。引物由捷瑞生物公司合成:P erm F(5′-GCAGGTCCAGCCCGACCCGAGC-3′)和P erm R(5′-GGTACCGTCGATCCTACCAACCGG-3′)用于扩增P erm E*启动子。
1.2 链霉菌的转化
操作方法参见文献[8]。
1.3 p NW-S2质粒的构建
大肠杆菌-链霉菌穿梭质粒p NW-S1中含lacZ启动子,在pNW-S1 lacZ诱导型启动子下游的转录起始位点和SD序列之间插入多克隆位点(Xba I、Spe I和Sca I),构建pNW-S2质粒,用于插入组成型启动子。
1.4 组成型质粒p NW-S3和p NW-S4的构建
以pIJ4090质粒为模板,通过PCR扩增后获得启动子P erm E*,与p NW-S2质粒分别进行XbaⅠ和SpeⅠ酶切后再通过T4连接酶16℃连接,转化入DH5α,重组质粒命名为p NW-S3。
p GH-P Sau3A通过Xba I、Spe I双酶切、胶回收后连接入p NW-S2质粒中,转化入DH5α后,重组质粒命名为p NW-S4。
1.5 PSS重组表达质粒的构建及PSS基因的亚克隆
根据p NW质粒的多克隆位点,设计PSS的上下游引物(Bam H IF:5′-CG GGATCC TTGATCAAGGTCGGAGGCGTGG-3′;Hin dⅢR:5′-CCC AAGCTT TCAGCCCTGGCAGAGGCCGCGG-3′),通过酶切连接的方法将PSS序列分别插入p NW-S3和p NWS4表达载体的Bam H I和Hin dⅢ之间,构建PSS重组表达质粒将构建的PSS重组表达质粒以及空载体p NW-S1通过PEG介导的原生质体法转化进入变铅青链霉菌TK24,铺抗生素水膜2 d后,长出大量白色菌体,挑取抗性转化子,分别提取其基因组,用PSS特异性引物扩增,得到目的基因1600 bp片断的阳性转化子。
1.6 磷脂酰丝氨酸合成酶的活性测定
酶活的测定采用酶联法。在文献[9]以及前期研究基础上,确定反应体系为:100μL 8 mmol·L-1底物+400μL底物缓冲溶液+10μL酶液(发酵上清液), 37℃反应10 min,加200μL EDTA反应终止液,煮沸5 min,加100μL 4-ATT溶液、10μL胆碱氧化酶, 5μL过氧化氢酶,37℃反应3 h,505 nm测OD值。
2 结果与讨论
2.1 p NW-S2质粒的构建
以p NW-S1质粒为模板,用相应引物分别PCR扩增插入多克隆位点上游1871 bp的F片段(图1a)和下游约400 bp的R片段(图1b),进行DNA电泳后胶回收,与p NW-S1质粒分别双酶切(图1c)后,进行三片段的连接后转化入DH5α。培养14 h后,长出均匀的单个克隆,转接入LB液体试管中,培养过夜后,送测序公司测序,结果显示阳性克隆已经成功插入3个单一的酶切位点ScaⅠ、SpeⅠ、XbaⅠ,p NW-S2构建成功。
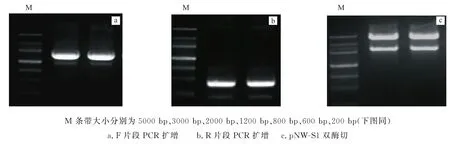
图1 pNW-S2质粒的构建Fig.1 Construction of plasmid pNW-S2
2.2 p NW-S3和p NW-S4质粒的构建
p IJ4090质粒用通用引物PCR扩增出约2000 bp的特异性引物,送测序公司测序后,返回结果与NCBI数据库的红霉素抗性启动子比对,该序列含有红霉素抗性基因的P erm E*启动子,通过设计的特异性引物P erm F和P erm R,PCR扩增得到P erm E*片段(图2a),切胶回收。同时,以p GH-P Sau3A质粒为模板, PCR扩增得到约130 bp的特异性条带(图2b),切胶回收。

图2 P ermE*启动子(a)、P Sau3A启动子(b)的PCR扩增Fig.2 PCR Amplification of the promoters P ermE*(a)and P Sau3A(b)
扩增得到的P erm E*和P Sau3A启动子片段与质粒p NW-S2分别进行SpeⅠ、XbaⅠ双酶切纯化后,连接,转化入DH5α后,挑取单个克隆进行PCR验证。PCR扩增时由于引物长度过小,不易与引物二聚体分开,故使用启动子上游引物和R片段下游引物进行PCR扩增。验证结果见图3,选取克隆1、4、8、12进行测序验证。根据测序返回结果,克隆1和8分别正确插入P erm E*和P Sau 3A片段,命名为p NW-S3、p NW-S4。

图3 p NW-S3和pNW-S4阳性克隆的PCR验证Fig.3 PCR Verification of positive clones p NW-S3 and p NW-S4
2.3 p NW-S系列质粒的构建(图4)

图4 p NW-S系列质粒的构建Fig.4 Construction of p NW-S plasmids
2.4 PSS基因的亚克隆
以p MD19T-PSS质粒为模板PCR扩增得到大小约1800 bp的PSS片段,与p NW-S3、p NW-S4质粒同时进行Bam H I、Hin dⅢ酶切,分别连接转化,最终筛选得到了p NW-S3-PSS和p NW-S4-PSS重组质粒。按1.5方法进行PSS基因的亚克隆,其PCR验证结果见图5。

图5 重组菌株pNW-S3-PSS和pNW-S4-PSS的PSS基因的PCR验证Fig.5 PCR Verification of PSSgene in pNW-S3-PSSand pNW-S4-PSS
2.5 启动子的活性分析
2.5.1 PSS的酶活
将重组克隆接入TBS液体培养基中培养3 d后,取各克隆发酵上清液测定PSS的酶活和蛋白总量,共进行4次发酵平行实验,结果取平均值,见表1。

表1 PSS酶活和蛋白总量Tab.1 PSS Activity and total protein
由表1可知,p NW-S3-PSS和p NW-S4-PSS的平均酶活分别达到40.2 U·m L-1和31.3 U·m L-1,是原始菌株PSS活性的31倍和23倍。P erm E*启动的重组菌p NW-S3-PSS的PSS酶活和蛋白总量高于P Sau3A启动的p NW-S4-PSS重组菌,说明P erm E*启动子的启动效果优于P Sau3A启动子。
2.5.2 PSS的蛋白表达
PSS在变铅青链霉菌TK24中的表达见图6A。

图6 PSS在变铅青链霉菌TK24中的表达Fig.6 The expression of PSSin Streptomyces lividans TK24
由图6A可以看出,p NW-S3-PSS和pNW-S4-PSS重组克隆的发酵上清液中均有明显的PSS目的条带,分子量与原始PSS的条带大小一致,约54 k Da,证明PSS在变铅青链霉菌TK24中获得成功表达。重组菌株p NW-S3-PSS发酵上清液中的PSS目的蛋白量明显高于p NW-S4-PSS,这与PSS酶活测定的结果一致,再次说明P erm E*的启动效果好于P Sau3A。同时在传代20次后,提取TK24的基因组,用PSS特异性引物PCR扩增,均扩增出一条约1600 bp的特异性条带(图6B),验证了PSS基因的存在,并且显示了这两个重组质粒的稳定性。
2.6 讨论
在所有红霉素抗性启动子中,P erm E*强启动子效果最好,它是在P erm E基础上将-35I区的TGG缺失,启动子活性大幅提高;启动子P Sau3A的-35区和-10区之间间隔18 bp,符合原核生物强启动子的最佳间隔(17±1)bp。并且由于这两个启动子都来源于链霉菌,所以在同源的链霉菌表达系统中,能够充分发挥启动子的作用,高效地启动表达外源基因,特别是同源基因。因此这两个启动子均能高效启动报道基因PSS的表达,最终PSS在变铅青链霉菌TK24中获得稳定表达,其表达活性比原始菌株的活性提高了20倍以上。同时,实验发现P erm E*启动PSS的效果要好于P Sau3A启动子。这可能是由于P erm E*启动子包含了2个-35区和-10区,属于多重启动子,有2个RNA聚合酶结合位点,而P Sau3A启动子片段较短,只含有1个-35区和-10区,只有1个RNA聚合酶结合位点,转录目的基因的效率略低于多重启动子。
通过对实验室已构建的诱导型链霉菌表达质粒p NW-S1进行改造,分别插入2个高效的组成型启动子P erm E*和P Sau3A,构建2个组成型链霉菌表达质粒p NW-S3和p NW-S4。PSS酶活和SDS蛋白电泳条带分析均表明PSS蛋白在变铅青链霉菌TK24中获得高效的表达。证明P ermE*和P Sau3A启动子在不需要诱导剂的条件下,成功启动了PSS基因的表达,链霉菌组成型表达质粒构建成功。同时,由于组成型启动子与诱导型启动子lacZ的并存(组成型启动子和诱导型启动子联用的启动效果正在进一步研究中),使得该组质粒可以在组成型表达和组成型-诱导型表达之间灵活选择,从而满足复杂多样的外源基因表达和工业生产要求,具有广阔的应用前景。
3 结论
红霉素抗性启动子P erm E*和来源于链霉菌核基因组片段的P Sau3A启动子均属于链霉菌组成型强启动子,分别将它们插入大肠杆菌-链霉菌穿梭质粒p NW-S1的SD序列和转录起始位点之间,构建成两个稳定的组成型表达质粒:p NW-S3和p NW-S4。以磷脂酰丝氨酸合成酶基因PSS为报道基因,以蛋白表达水平评价这两个启动子在变铅青链霉菌TK24中的表达效果。结果表明,在两株组成型表达重组菌中,PSS均得到了高效表达,具有在链霉菌系统中高效表达外源基因的功能,其中P erm E*的启动效率略高于P Sau3A。
[1] 王丽非,洪斌.变铅青链霉菌TK24 secE启动子表达特性的研究[J].遗传学报,2003,30(4):370-375.
[2] 李佳,向四海,杨秀山,等.报告基因法比较两种放线菌启动子的活性[J].微生物学报,2009,49(11):1454-1458.
[3] 范与庆,闵勇,熊伟,等.orfX基因整合表达在阿维链霉菌工业育种中的应用[J].农业生物技术学报,2007,15(6):1048-1052.
[4] 何群香,闵勇,王华丽,等.提高耐热链霉菌转化泰乐菌素能力的分子改造与育种[J].农业生物技术学报,2008,16(6):1012-1018.
[5] Horinouchi S,Beppu T.Construction and application of a promoterprobe plasmid that allows chromogenic identification in Streptomyces lividans[J].Journal of Bacteriology,1985,162(1):406-412.
[6] Horinouchi S,Nishiyama M,Nakamura A,et al.Construction and characterization of multicopy expression-vectors in Streptomyces spp. [J].Mol Gen Genet,1987,210(3):468-475.
[7] Hopwood D A,Bibb M J,Chater K F,et al.Genetic Manipulation of Streptomyces.A Laboratory Manual[M].Norwich:John Innes Foundation Press,1985:84-170.
[8] 吴胜,夏焕章,程杉.黑暗链霉菌原生质体制备、再生及其DNA转化条件的研究[J].沈阳药科大学学报,2001,18(3):213-216.
[9] Imamura S,Horiuti Y.Enzymatic determination of phospholipase D activity with choline oxidase[J].Journal of Biochemistry,1978, 83(3):677-680.
Promotion Efficiency Comparison of Constitutive Promoters P ermE*and P Sau3A in Streptomyces Lividans
ZHANG Ying,TAO Xin-yi,WANG Feng-qing,WEI Dong-zhi
(National Engineering Research Center for Biotechnology,Newworld Institute of Biotechnology, East China University of Science and Technology,Shanghai 200237,China)
Erythromycin resistance promoter P erm E*and promoter fragment P Sau3A from Streptomyces nuclear genome are constitutive promoters stronger than general promoter.They were inserted between the SD sequence and transcription start site of the E.coli-Streptomyces shuttle plasmid p NW-S1 to construct two stable constitutive expression plasmids:p NW-S3 and p NW-S4.Phosphatidylserine synthase(PSS)gene,as a reporter gene,was cloned into plasmid p NW-S3 and p NW-S4 to be transformed into Streptomyces lividans TK24.The promoter activity was evaluated by SDS electrophoresis and PSS activity assay.It showed that PSS was over-expressed with 20 times activity compared to wild strain in TK24,which proved the p NW-S3 and p NW-S4 expression vectors could highly express foreign genes.Promotion effeciency of P erm E*was superior to that of P Sau3A.
constitutive promoter;Streptomyces;phosphatidylserine synthase
Q 785
A
1672-5425(2013)11-0047-04
10.3969/j.issn.1672-5425.2013.11.011
2013-09-10
张莹(1987-),女,陕西汉中人,硕士研究生,研究方向:生物化学与分子生物学;通讯作者:魏东芝,教授,E-mail:dzhwei @ecust.edu.cn。
