TiO2纳米管阵列电极电催化降解甲基橙的研究
2013-08-16张金涛刘宝亮徐晓羚
张金涛,刘宝亮,徐晓羚
(常州工学院化工系,江苏常州213022)
TiO2纳米管阵列电极电催化降解甲基橙的研究
张金涛,刘宝亮,徐晓羚
(常州工学院化工系,江苏常州213022)
采用阳极氧化法制备了TiO2纳米管阵列电极。采用场发射扫描电子显微镜(FESEM)和X-射线衍射仪(XRD)表征了电极的表面形貌和晶体结构,应用电化学阻抗谱(EIS)技术研究了不同阳极极化电位下电极的导电性能,应用荧光光谱法研究了电极产生羟基自由基(·OH)的活性,考察了3种支持电解质(Na2SO4、Na NO3和NaCl)对甲基橙(MO)电催化降解效率的影响,通过加入捕获剂探讨了MO分子的降解机理。结果表明,TiO2纳米管阵列电极电极的导电性随阳极极化电位的升高而增强;在电场作用下,TiO2纳米管阵列电极表面生成大量·OH;Na2SO4和NaNO3不参与MO分子的氧化反应,MO的降解符合一级反应动力学模型,而NaCl参与了TiO2纳米管阵列电极电催化降解MO的过程,呈现出复杂的动力学行为;在捕获剂存在的情况下,MO分子仍能发生降解,显示MO分子可在TiO2纳米管阵列电极表面直接氧化。
TiO2纳米管阵列电极;甲基橙;电催化降解;羟基自由基
TiO2纳米管的特殊结构,使其具有更大的比表面积和更强的吸附能力,作为光催化材料其性能显著优于粉态TiO2纳米材料[1,2]。TiO2纳米管的制备通常采用水热合成法、模板法和阳极氧化法等。其中,阳极氧化法具有成本低、操作简便易行、所得TiO2纳米管分布均匀且与基体结合牢固等优点而倍受研究者的青睐[3-7],并很好地解决了纳米TiO2粉末难于回收再利用的难题[3]。近年来,研究人员广泛而深入地研究了阳极氧化法制备参数对TiO2纳米管阵列电极的光催化性能的影响[5-7]及通过掺杂元素[8,9]、施加阳极偏压等方式[10]提高TiO2纳米管阵列电极光催化性能的可行性。
与此同时,应用TiO2电极电催化降解有机污染物的研究进展很大。张翼等[11]采用溶胶-凝胶法在钛金属表面制备铈掺杂TiO2纳米薄膜,并以此为电极进行了油田废水的电催化降解研究。结果表明,施加16.5 V的直流电压时,该电极能够有效地降低油田废水的COD。刘弋潞等[12]在不锈钢基体上采用溶胶-凝胶法制备了TiO2纳米薄膜,讨论了电解电压、电极间距、p H值、电解时间以及电解质(NaCl)等对印染废水电催化降解效果的影响。结果表明,该电极具有优异的电催化氧化性能。刘贵昂等[13]采用复合电镀法在泡沫铝上制备了纳米TiO2涂层,并以此为电极,采用12 V直流电压进行了电催化降解次甲基蓝的研究。结果表明,泡沫铝负载纳米TiO2的电催化活性明显优于其光催化活性。本课题组在前期工作中,初步研究了TiO2纳米管阵列电极电催化降解甲基橙(MO)的活性及其影响因素,并探讨了MO降解反应动力学。结果表明,MO溶液p H值、电解槽压、支持电解质浓度等是影响TiO2纳米管阵列电极电催化降解效率的重要因素,MO电催化降解反应遵循一级反应动力学规律。
作者在此采用阳极氧化法在钛箔表面制备TiO2纳米管阵列电极,采用场发射扫描电子显微镜(FESEM)和X-射线衍射仪(XRD)表征电极的表面形貌和晶体结构;并以其作为阳极进行模拟染料废水(MO溶液)的电化学氧化,以对苯二甲酸为探针,采用荧光光谱法测定电化学氧化过程中电极产生的羟基自由基(·OH),考察支持电解质和捕获剂对电催化降解效率和降解过程的影响,探讨了MO的电催化降解机理。
1 实验
1.1 试剂与仪器
TA1工业纯钛箔(100 mm×30 mm×0.2 mm),宝鸡高新区国星薄材加工厂;甲基橙、硝酸、硫酸、氢氧化钠、OP乳化剂,丹阳永丰化学试剂厂;氢氟酸(HF),上海申博化工有限公司;磷酸钠,上海新华化工厂;无水硫酸钠、氟化钠、氯化钠、硅酸钠、无水碳酸钠、叔丁醇,国药集团上海化学试剂有限公司;对苯二甲酸,上海润捷化学试剂有限公司;除OP乳化剂和叔丁醇为化学纯外,其余试剂皆为分析纯。
UltimaⅣ型X-射线衍射仪,日本Rigaku公司; DWW-K型直流电源,扬州格尔仕电源科技有限公司; KSL-1100X型箱式电阻炉,合肥科晶材料技术有限公司;UV1102型紫外可见分光光度计,上海天美科学仪器有限公司;721E型紫外可见分光光度计,上海光谱仪器有限公司;DF-101S型集热式恒温加热磁力搅拌器,上海越众仪器设备有限公司;S4800型场发射扫描电子显微镜、F-4600型荧光光谱仪,日本日立公司。
1.2 TiO2电极的制备
将TA1工业纯钛箔用自制除油液(Na3PO420 g ·L-1,Na2SiO34 g·L-1,OP乳化剂3 g·L-1)于55℃清洗2 min,再用HF和HNO3混合溶液(40% HF∶浓HNO3∶H2O为1∶4∶5,体积比)[14]浸蚀,然后用去离子水清洗,晾干备用。以钛箔为阳极、两片304不锈钢片为阴极,置于240 m L 0.5%(质量分数) HF水溶液(恒温水浴16℃)中进行阳极氧化,极间距为2 cm,直流电压为20 V;在电磁搅拌下阳极氧化25 min后立即取出钛箔,用大量去离子水冲洗并晾干,置于箱式电阻炉中以10℃·min-1升温至350℃保持3 h,随炉冷却至室温。
1.3 羟基自由基的荧光检测
参照文献[15]配制0.5 mmol·L-1对苯二甲酸溶液(含0.25 mol·L-1硫酸钠、2.0 mmol·L-1NaOH)。以TiO2纳米管阵列为阳极、两片相同面积的钛板为阴极,极间距为2 cm,直流电压为10 V,溶液体积为200 m L(恒温水浴20℃),电极浸入面积均约为24 cm2。每隔10 min取样2 m L,稀释25倍,在荧光光谱仪上测定2-羟基对苯二甲酸的荧光强度,激发波长λex=315 nm,发射波长λem=424 nm。
1.4 支持电解质的影响及MO降解反应动力学研究
为研究支持电解质对电极电催化降解效率的影响规律,选用3种钠盐(Na2SO4、NaNO3和NaCl)为电催化降解MO的支持电解质,浓度均为0.035 mol· L-1。如无特别说明,MO体系均如下:MO溶液浓度10 mg·L-1,体积200 m L,p H=3,溶液温度20℃。电解装置同1.3。在0~25 mg·L-1浓度范围内,MO的吸光度与其浓度呈现非常好的线性关系,因而在MO的催化降解过程中,可以吸光度代替浓度进行MO电催化反应动力学的探讨。每隔一定时间取样,测定MO溶液的吸光度,按式(1)计算脱色率R。

式中:A0为初始溶液的吸光度值;At为降解后溶液的吸光度值。
1.5 电极表面形貌和晶体结构
TiO2纳米管阵列电极的表面形貌采用场发射扫描电子显微镜观察,加速电压为15 k V,照片放大倍率为50 000。
TiO2纳米管阵列电极的晶体结构采用X-射线衍射仪分析,测试条件:Cu靶,10°·min-1,管压40 k V,电流30.0 m A,波长0.154059 nm。
1.6 电化学阻抗谱(EIS)测试
TiO2纳米管阵列电极的EIS测试采用经典三电极体系,其中TiO2纳米管阵列电极为工作电极、铂片为对电极、218型Ag/AgCl电极为参比电极。支持电解质为0.5 mol·L-1的硫酸钠溶液,试样测试面积约为3.8 cm2。EIS测试在CHI660B电化学工作站(上海辰华仪器公司)上进行,测试软件为仪器随机附带电化学综合测试系统。测试在开路电位和多种阳极极化电位下完成,施加振幅为10 m V的正弦波电位扰动,测试频率范围为105~10-2Hz。所有测试均在室温下进行。
2 结果与讨论
2.1 TiO2纳米管阵列电极的表征
对热处理后的TiO2纳米管阵列电极的表面形貌和晶体结构进行了表征,其SEM照片和XRD图谱见图1。
由图1a可知,阳极氧化和热处理后,钛基体表面生成了基本均匀、高度有序的TiO2纳米管阵列,纳米管内径为100 nm左右。由于纳米管具有中空结构及存在管间隙导致其拥有巨大的比表面积,因而具有更强的吸附有机污染物分子的能力,为电催化降解污染物提供了良好的物质基础。由图1b可知,经过350℃热处理后,2θ为25.3°(101晶面)和48°(200晶面)两处呈现锐钛矿+++++++型TiO2的特征衍射峰(JCPDS 21-1272),且25.3°(101晶面)衍射峰的强度较大,表明此温度下无定型TiO2已经完全转化为晶体。同时发现,钛基体衍射峰的强度很大,可能是由于生成的TiO2纳米管阵列厚度较小的缘故。
2.2 TiO2纳米管阵列电极的EIS分析
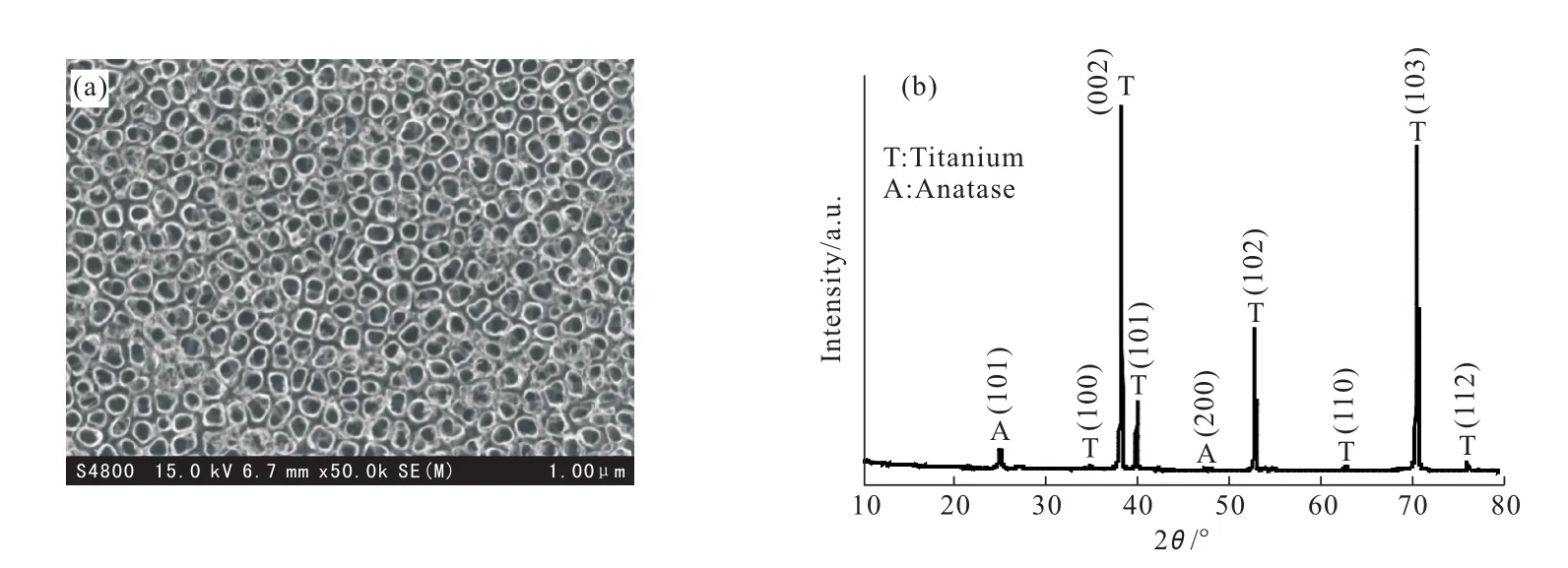
+图1 TiO2纳米管阵列电极的SEM照片(a)和XRD图谱(b)Fig.1 SEM Image(a)and XRD pattern(b)of TiO2nanotube array electrode
电化学阻抗谱(EIS)技术更多地应用于金属腐蚀与防护方面的研究[16,17]。在不破坏电极样品表面状态的条件下,能够获得电极/溶液界面的双电层电容、电极反应的电荷转移电阻等参数,对于了解电极的腐蚀过程具有重要的意义。对于电化学阻抗谱,在相同频率下,阻抗值越大,电荷在电极/溶液界面或保护层内部的传递越困难,表明电极反应越不易于发生。为研究TiO2纳米管阵列电极在电催化降解有机污染物过程中的导电性能及外加电压对其催化性能的影响,对TiO2纳米管阵列电极进行了EIS测试。分别测定TiO2纳米管阵列电极在开路电位(OCP)及不同阳极极化电位下的EIS曲线,结果见图2。其中,图2a和图2b为Nyquist图,图2c和图2d分别为阻抗Bode图和相位角Bode图。

图2 TiO2纳米管阵列电极在开路电位(OCP)和各阳极极化电位下的EIS曲线Fig.2 Nyquist plots(a,b),impedance-Bode(c)and phase angle-Bode(d)curves of TiO2nanotube array electrode at different anode polarization potentials
由图2a和图2b可知,TiO2纳米管阵列电极在开路电位(OCP)下的Nyquist曲线为一个半径非常大的圆弧,表明此时的电极具有较大的电荷转移电阻[18,19],不利于电催化氧化有机污染物;随着阳极极化电位的升高,Nyquist曲线的半径显著减小,表明电极的电阻明显降低。同时发现,在不同阳极极化电位下,Nyquist曲线均呈现出多个时间常数,低频区近乎直线,意味着溶液中离子在电极表面层中表现为扩散传输[20]。
Nyquist曲线无法详尽给出电极在全部频率范围内阻抗变化的形态,而通过电极体系的阻抗Bode图(图2c)和相位角Bode图(图2d)能够更清晰地了解到阳极极化电压对电极导电性能的影响效果。在开路电位下测得的电极EIS曲线呈现为两个特征区域:高频区(100~1 k Hz)阻抗为恒定值,相应的相位角接近于零,此对应于溶液电阻;中低频区阻抗随频率的降低而增大,呈现为斜率几乎为-1的直线,相应的相位角形成一个接近于-90°的平台,此对应于TiO2纳米管阵列膜的电容行为。
此外,由图2d可了解TiO2纳米管阵列电极的结构:在103~10-1Hz频率区域内,存在两个时间常数,表明TiO2纳米管阵列电极由纳米管层和致密内层两部分构成[21]。在施加3.0~9.0 V阳极极化电位的情况下,电极的EIS曲线表现出3个区域:高、中频区阻抗和相位角的形态与OCP时相似;低频区阻抗值下降并趋于恒定,且随阳极极化电位的增大数值显著减小,而相位角平台宽度随阳极极化电位的增大逐步收缩。这些变化均表明TiO2纳米管阵列电极的导电性能受到阳极极化电位的显著影响,极化电位越高电极的导电性越好[22]。
通过阻抗分析可知,TiO2纳米管阵列电极的总阻抗随阳极极化电位的增大而迅速减小,此意味着在以TiO2纳米管阵列为阳极进行电催化降解有机物时,采用10 V槽电压可有效降低电极的电阻,形成良好的导电状态,为TiO2纳米管阵列电极直接或间接氧化降解有机污染物提供物质基础。
2.3 羟基自由基的荧光检测
研究表明[23],有机物在金属氧化物电极作用下的电催化降解可能存在不同方式:直接氧化、间接氧化或二者同时发生。在以TiO2为催化剂的光催化或光电催化降解有机物的研究中,通常认为光生空穴及由此产生的羟基自由基(·OH)是导致有机物降解的主要物种[24-27]。对苯二甲酸可捕获羟基自由基生成具有荧光性质的2-羟基对苯二甲酸[15]。2-羟基对苯二甲酸的荧光强度与其浓度成正比,可以其荧光强度代表浓度进行探讨[15]。因此,以对苯二甲酸为捕获剂,采用荧光光谱法考察TiO2纳米管阵列电极在电催化降解有机污染物的过程中产生羟基自由基的情况。
2-羟基对苯二甲酸的荧光强度随电解时间的变化曲线见图3。

图3 对苯二甲酸溶液的荧光光谱(a)及424 nm处荧光强度随电解时间的变化曲线(b)Fig.3 Fluorescence spectra of terephthalic acid solution(a)and its change curves of fluorescence intensity at 424 nm with electrolyze time(b)
由图3a可知,对苯二甲酸溶液在电解前没有任何荧光性质。电解开始后,由于对苯二甲酸捕获了电极产生的羟基自由基,生成了具有荧光性质的2-羟基对苯二甲酸,在约424 nm处产生荧光信号,并随着电解的进行,该信号的强度快速增大。由图3b可知,在全部电解时间内,羟基自由基的生成速率并不完全相同。电解前70 min,羟基自由基的生成速率较快。羟基自由基生成反应符合零级反应动力学规律[15],拟合可得其表观速率常数为16.92 min-1,相关系数为0.9871;电解70 min后的生成速率有一定程度的降低。可能是由于电解消耗了对苯二甲酸,导致羟基自由基与其反应减弱,生成的具有荧光性质的2-羟基对苯二甲酸的量减少。上述结果表明,TiO2纳米管阵列电极具有很好的产生羟基自由基的能力。同时,也说明部分MO分子在电解过程中可能是通过TiO2纳米管阵列电极生成的羟基自由基的强氧化作用而发生降解。
2.4 捕获剂对MO降解的影响
叔丁醇[25,26]和柠檬酸[27,28]是TiO2光催化或光电催化反应中常用的光生空穴或羟基自由基的捕获剂,当它们存在于反应体系中时,可对有机物分子的降解效率产生重大影响。碳酸钠[29]作为羟基自由基的抑制剂,在金属氧化物电极(如Ti/Pb O2电极)的电化学催化反应中对羟基自由基具有非常显著的抑制作用。F-可强烈吸附在TiO2催化剂表面,与有机物分子在催化剂表面的吸附形成竞争,并取代催化剂表面吸附的羟基自由基[25,26],导致有机物分子的降解显著受阻。因此,为研究MO分子的降解是发生在TiO2纳米管阵列电极表面的直接氧化还是通过TiO2纳米管阵列电极在电场作用下产生的羟基自由基在溶液中对MO分子氧化,在MO体系中分别加入浓度均为0.05 mol·L-1的叔丁醇、碳酸钠和氟化钠溶液,调节p H= 11±0.02,考察MO的降解效果,结果见图4。
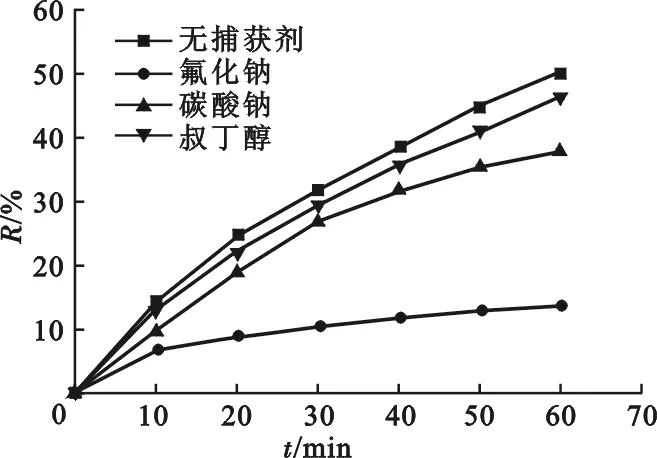
图4 捕获剂对MO降解率的影响Fig.4 Effect of capture agents on electrocatalytic degradation efficiency of MO on TiO2nanotube array electrode
由图4可知,3种捕获剂的引入均导致MO体系脱色率下降,尤以氟化钠的效果最为显著。而叔丁醇仅仅导致MO脱色率发生很小的变化,在全部电解时间内与无捕获剂MO体系的脱色率比较接近。
文献[25,26]指出,叔丁醇对TiO2光(电)催化降解有机物分子的效率具有重要影响,其对溶液中游离羟基自由基的捕获能力极强,导致溶液中可与有机物分子反应的游离羟基自由基数量急剧减少,从而显著降低有机物分子的降解效率。但本研究中叔丁醇的加入并未严重影响MO分子在TiO2纳米管阵列电极作用下的降解,60 min后MO脱色率仅由加入前的约51%下降至约46%。这可能是由于在电催化体系中叔丁醇分子在电极表面发生降解所致,同时也表明MO分子的降解可能不仅是通过与溶液中羟基自由基的作用来进行的,还应包含电极表面直接氧化的过程。
在相同条件下,由于CO2-3在电极表面可能不再发生电化学反应,可很好地捕获羟基自由基或空穴,有效减少了MO与羟基自由基或空穴反应的机会,从而导致MO分子降解率降低较明显。尽管CO2-3的存在较大程度上影响了MO的降解,但MO分子在较长时间电解后仍然发生了一定程度的降解,60 min后脱色率约为38%,达到无捕获剂MO体系脱色率的3/ 4,表明部分MO分子可能在电极表面发生了直接电化学氧化反应而发生破坏。
氟化钠引入后,由于F-可强烈吸附于电极表面,阻碍了羟基自由基在电极表面的吸附,进而导致MO分子无法与其反应,从而导致溶液脱色率显著下降; 60 min后MO脱色率约为14%,仅为无捕获剂MO体系脱色率的1/4。在F-有效夺取电极表面羟基自由基的吸附位点后,仍有一部分MO分子发生电催化降解反应,表明该反应为电极直接氧化行为。
综上所述,MO分子在TiO2纳米管阵列电极作用下的电催化降解行为应包含MO分子在电极表面的直接氧化和由羟基自由基引起的间接氧化两种过程。
2.5 支持电解质对电催化降解效率的影响及反应动力学
在传统电化学过程中,为增强有机物溶液的导电性,通常需加入一定浓度的支持电解质。选择Na2SO4、Na NO3和NaCl作为支持电解质,研究它们对TiO2纳米管阵列电极电催化降解MO的影响,结果见图5。
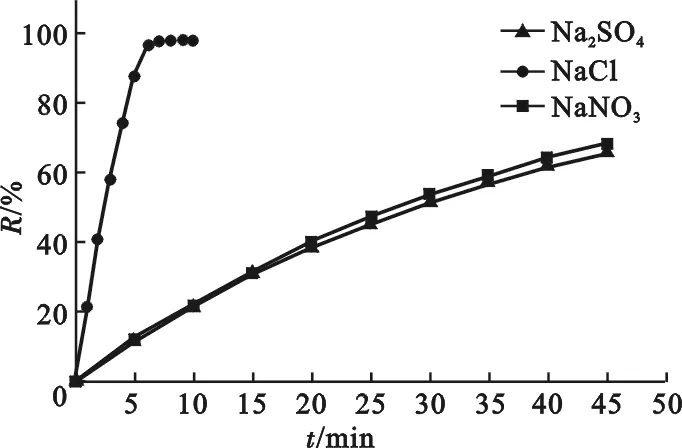
图5 支持电解质对电催化降解MO效率的影响Fig.5 Effect of supporting electrolytes on electrocatalytic degradation efficiency of MO on TiO2nanotube array electrode
由图5可知,以NaCl为支持电解质的MO体系脱色极为迅速,在10 min内脱色率达98.3%;而Na2SO4和Na NO3作为支持电解质的两种MO体系脱色效果远低于NaCl,45 min时二者的脱色率仅分别为65.8%和68.2%。由于Na2SO4与NaNO3对MO电催化降解效率的影响非常相近,可认为Na2SO4与Na NO3在此只是发挥了增强MO溶液导电性的作用。为验证Na2SO4、NaNO3和NaCl三种支持电解质在电催化降解MO过程中发挥的作用存在差异,测定了TiO2纳米管阵列电极电催化降解MO的动力学曲线,见图6。
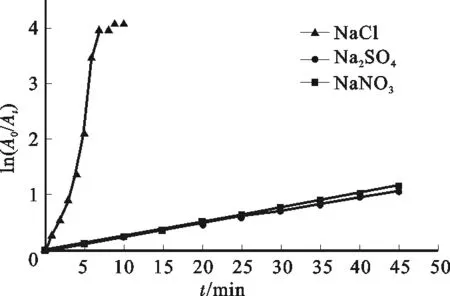
图6 MO电催化降解的动力学曲线Fig.6 Kinetic curves of MO electrocatalytic degradation on TiO2nanotube array electrode
通过对图6数据进行线性拟合,得到以Na2SO4和NaNO3作为支持电解质的两种MO溶液体系的动力学曲线相关系数均为0.9998,表明TiO2纳米管阵列电极电催化降解MO的反应符合一级反应动力学,降解反应的表观速率常数分别为0.02394 min-1(Na2SO4)和0.02567 min-1(NaNO3)。MO分子的TiO2电催化降解反应动力学与TiO2光催化降解反应动力学一致,均具有一级反应特征[1]。按一级反应动力学方程获得的以NaCl为支持电解质的MO溶液降解的ln(A0/At)~t关系呈现S形,表明该体系中MO分子的降解过程中应有其它活性物质参与。这与文献[28]的分析一致:NaCl作为支持电解质的MO体系,电解过程中Cl-可能被氧化生成Cl2、HCl O、Cl O-等强氧化性物质,MO分子在这些强氧化性物质作用下发生结构破坏,从而导致其脱色。
为验证NaCl与Na2SO4和Na NO3的作用不同,测定了三种电解质的MO体系电催化降解过程的紫外可见吸收光谱,结果见图7。

图7 分别以Na2SO4(a)、NaNO3(b)和NaCl(c)为支持电解质的MO溶液的紫外可见吸收光谱Fig.7 UV-Vis Spectra of MO solutions with Na2SO4(a),NaNO3(b)and NaCl(c)as supporting electrolytes,respectively
由图7可知,对于三种支持电解质,MO溶液的最大吸收峰均出现在503 nm处,该吸收峰对应于分子中的醌式结构。而在315 nm和277 nm(图7a和图7b)、277 nm和270 nm(图7c)等处也具有一定强度的吸收,可能对应于MO分子中的苯环结构[30]。在经历一定时间的电解后,3种MO体系的可见最大吸收峰几乎消失,表明MO的结构遭到破坏,发色基团已经降解;且NaCl作为支持电解质更有利于MO的电催化降解,其脱色效率远高于另两种支持电解质。
随电解的进行,图7a和图7b中紫外吸收峰(315 nm和277 nm处)显著减弱,表明MO分子在TiO2纳米管阵列电极作用下被破坏,生成了其它小分子(可能是无色有机小分子或进一步氧化生成的CO2和H2O等);图7c中315 nm处紫外吸收峰显著降低,277 nm处的吸收峰发生蓝移(约7 nm),产生了新的吸收峰,并呈现出随电解的进行先增大后减小的趋势,继续进行电解时,该处吸收峰强度进一步降低。推测可能是由于电解过程中MO的降解产物导致该处吸收峰的出现。关于该吸收峰对应于何种有机物分子,有待进一步研究。
3 结论
采用阳极氧化法在氢氟酸水溶液中制备了TiO2纳米管阵列电极,对其电催化降解MO的性能进行了研究。
(1)EIS分析表明,TiO2纳米管阵列电极的阻抗随阳极极化电位的增加而显著降低,在较高的槽电压下TiO2纳米管阵列电极具有良好的导电性。
(2)TiO2纳米管阵列电极具有较高的产生羟基自由基的活性。MO在反应体系中以直接氧化与间接氧化协同方式进行氧化降解。
(3)在以Na2SO4和NaNO3为支持电解质的体系中,MO降解反应符合一级反应动力学规律。而在以NaCl为支持电解质的体系中,MO的降解反应呈现出复杂的机理。
[1] 庄惠芳,赖跃坤,李静,等.高度有序的二氧化钛纳米管阵列的制备及其光催化活性的研究[J].化学学报,2007,65(21):2363-2369.
[2] 刘海津,刘国光,侯泽华,等.电化学方法制备掺杂二氧化钛纳米管阵列[J].稀有金属材料与工程,2011,40(4):723-727.
[3] 陈秀琴,张兴旺,雷乐成.自组织TiO2纳米管阵列的制备及形成机理[J].中国有色金属学报,2010,20(9):1724-1731.
[4] Xie Y B,Zhou L M,Lu J.Photoelectrochemical behavior of titania nanotube array grown on nanocrystalline titanium[J].Journal of Materials Science,2009,44(11):2907-2915.
[5] 李洪义,王金淑,陈欣,等.TiO2纳米管阵列薄膜制备及生长机理的研究[J].无机化学学报,2010,26(2):217-222.
[6] Alivov Y,Fan Z Y,Johnstone D.Titanium nanotubes grown by titanium anodization[J].Journal of Applied Physics,2009,106(3): 034314-034318.
[7] Kang X W,Chen S W.Photocatalytic reduction of methylene blue by TiO2nanotube arrays:Effects of TiO2crystalline phase[J]. Journal of Materials Science,2010,45(10):2696-2702.
[8] Hu K H,Hu X G,Xu Y F,et al.Synthesis of nano-MoS2/TiO2composite and its catalytic degradation effect on methyl orange[J].Journal of Materials Science,2010,45(10):2640-2648.
[9] Archana P S,Jose R,Jin T M,et al.Structural and electrical properties of Nb-doped anatase TiO2nanowires by electrospinning[J]. J Am Ceram Soc,2010,93(12):4096-4102.
[10] 王宁,李新勇,侯阳,等.TiO2纳米管的阳极氧化法制备及对对氯苯酚的光电降解研究[J].科学通报,2008,53(13):1528-1532.
[11] 张翼,于婷,张玉洁,等.铈掺杂Ti/TiO2电极的制备及催化降解油田废水性能[J].催化学报,2009,30(2):154-158.
[12] 刘弋潞,卢维奇,黄贵明.电催化氧化法处理印染废水的实验研究[J].化学与生物工程,2009,26(2):58-61.
[13] 刘贵昂,赖国霞.复合电镀法制备纳米TiO2涂层光催化及电催化特性的比较[J].钛工业进展,2009,26(6):35-38.
[14] 阮修莉,刘长峰,陈晓慧.TiO2纳米管电极光电催化降解五氯苯酚[J].水处理技术,2011,37(1):44-47.
[15] 丁海洋,冯玉杰,吕江维,等.钛基二氧化锡电极电解过程中羟基自由基检测及电催化机理[J].分析化学,2007,35(10):1395-1399.
[16] 张金涛,胡吉明,张鉴清,等.LY12铝合金/钝化膜/环氧涂层复合电极的腐蚀电化学行为[J].金属学报,2006,42(5):528-532.
[17] 张金涛,杨春勇,潘亮,等.2A12铝合金表面铈盐掺杂硅烷杂化膜在3.5%NaCl溶液中耐蚀性能的电化学研究[J].金属学报, 2008,44(11):1372-1377.
[18] 张溪,廖雷,凌云汉,等.氧气热处理的TiO2纳米管阵列光电催化降解亚甲基蓝的研究[J].环境科学,2011,32(11):3372-3378.
[19] 张胜寒,梁可心,檀玉.电化学阻抗谱法研究铈改性TiO2纳米管阵列光电极裂解水产氢动力学[J].化学学报,2012,70(9):1109-1116.
[20] 田西林,陶杰,陶海军,等.阳极氧化制备TiO2纳米管阵列电极反应及阻抗研究[J].稀有金属材料与工程,2010,39(6):1066-1070.
[21] Fadl-Allah S A,El-Sherief R M,Badawy W A.Electrochemical formation and characterization of porous titania(TiO2)films on Ti[J].J Appl Electrochem,2008,38(10):1459-1466.
[22] Diamanti M V,Bolzoni F,Ormellese M,et al.Characterisation of titanium oxide films by potentiodynamic polarisation and electrochemical impedance spectroscopy[J].Corrosion Engineering,Science and Technology,2010,45(6):428-434.
[23] 史艳华,孟惠民,俞宏英,等.贵金属氧化物涂层电极催化降解苯胺[J].北京科技大学学报,2008,30(4):359-363.
[24] 魏凤玉,桑蕾.高光催化活性和易分离回收的硫掺杂TiO2纳米管[J].催化学报,2009,30(4):335-339.
[25] 金辰,邱顺晨,朱月香,等.具有优异热稳定性的磷修饰氧化钛及其对水中污染物的降解[J].催化学报,2011,32(7):1173-1179.
[26] 宋爽,洪甜蜜,方慧莹,等.可见光响应的碘掺杂TiO2光催化剂的制备及其作用机理[J].环境工程学报,2013,7(1):1-6.
[27] 丛燕青,李哲,张轶,等.Fe2O3/TiO2纳米管的制备及其光电催化降解染料废水性能[J].催化学报,2012,33(8):1402-1409.
[28] 汪青,尚静,宋寒.影响TiO2纳米管光电催化还原Cr(Ⅵ)的因素探讨[J].催化学报,2011,32(9):1525-1530.
[29] 吴迪.羟基自由基在电催化氧化体系中的形成规律及其在废水处理中的应用研究[D].长春:吉林大学,2007.
[30] 王娟,申婷婷,李小明,等.Fe(Ⅱ)EDTA/H2O2电催化降解甲基橙模拟废水的研究[J].环境工程学报,2010,4(4):833-838.
Electrocatalytic Degradation of Methyl Orange on TiO2Nanotube Array Electrode
ZHANG Jin-tao,LIU Bao-liang,XU Xiao-ling
(Department of Chemical Engineering,Changzhou Institute of Technology,Changzhou 213022,China)
TiO2Nanotube array electrodes were prepared using anodic oxidation method.The morphology and the crystal structure of TiO2nanotube array electrode were characterized by filed emission scanning electron microscope(FESEM)and X-ray diffraction(XRD),respectively.Using electrochemical impedance spectroscopy(EIS)technique,the difference of conductivity of TiO2nanotube array electrode was investigated under various anodic polarization potentials.The electrode activity for producing hydroxyl radicals(·OH)was studied by fluorescence spectrum technique using terephthalic acid as·OH capture agent.The effects of three kinds of supporting electrolytes were investigated on electrocatalytic degradation efficiency of MO.The degradation reaction mechanism of MO was discussed in the presence of·OH capture agent.The results indicated that the conductivity of TiO2nanotube array electrode increased with anodic polarization potentials.A great quantity of·OH generated on the surface of TiO2nanotube electrode in the presence of electric field.The two kinds of supporting electrolytes,Na2SO4and Na NO3,had not been involved in the oxidizing reaction of MO and the electrocatalytic degradation of MO accorded with the first order reaction kinetics by the action of TiO2nanotube array electrode.However,the supporting electrolyte of NaCl took effect to electrocatalytic degradation of TiO2nanotube array electrode for MO,leading to the complex reaction kinetics.In the presence of·OH capture agent,the degradation of MO still took place,suggesting that MO could be directly oxidized on the surface of TiO2nanotube array electrode.
TiO2nanotube array electrode;methyl orange;electrocatalytic degradation;hydroxyl radical
O 614.411 X 703
A
1672-5425(2013)11-0031-07
10.3969/j.issn.1672-5425.2013.11.008
常州市应用基础研究计划项目(CJ20120019)
2013-08-19
张金涛(1969-),男,山东阳谷人,博士,教授,研究方向:腐蚀电化学,E-mail:jintaozh2002@163.com。
