中国一进行性肌营养不良家系相关致病基因Dystrophin突变检测结果分析
2013-06-15朱元洲柯琴梅祝建芳
田 莉,张 凌,杜 戎,朱元洲,柯琴梅,祝建芳
(华中科技大学同济医学院附属协和医院,武汉430022)
进行性肌营养不良(DMD)是一种致死性的常见肌肉遗传疾病。根据目前研究,DMD发病多与遗传因素有关[1]。在众多确定与DMD发病有关的致病基因中,抗肌萎缩蛋白基因(Dystrophin基因)备受关注。Dystrophin基因位于X染色体短臂(Xp 21.1-3),是目前已知最长的人类基因。根据国外报道,其主要突变类型为基因部分缺失,占全部突变类型的60% ~65%[2~4]。本研究以Dystrophin基因为候选基因,对一个中国DMD家系成员进行了基因突变检测,以期了解Dystrophin基因突变情况。
1 资料与方法
1.1 临床资料 该DMD家系来自中国河南,其家系有1例确诊患者(先证者),家系图见图1。此患者为15岁男性,近半年出现进行性肌无力,无既往特殊病史和家族史,其血清肌酸激酶明显增高。我们对此家系的所有成员进行临床调查,包括是否出现肌无力和肌萎缩、肌酶变化、家族史等,并作全面肌酶检测。阳性患者符合以下诊断标准:①以缓慢进行性的对称性肢体近端肌萎缩和肌无力为主症,呈翼状肩胛、鸭步、肌病面容或假性肥大等征象,但无肌肉压痛,Gower征阳性;②多在儿童和青少年期发病,常有家族遗传史;③尿肌酸增加,肌酐减少,血清肌酸磷酸激酶和乳酸脱氢酶等增高,血、尿肌红蛋白增高;④肌电图可见自发电活动增多,轻收缩时显示多相波明显增多,电位时限缩短,波幅降低,并有病理干扰相;⑤肌肉活检可见肌纤维肿胀或萎缩、变性,大量脂肪和结缔组织增生。在获得家系成员知情同意后,抽取外周静脉血5 mL,置于含有ACD抗凝管中备用。随机抽取140例家系外正常健康对照者,其中男64例,女76例,均无肌萎缩及肌无力表现,无肌酶改变,获得知情同意后,抽取外周静脉血5 mL,置于含有ACD的抗凝管中备用。
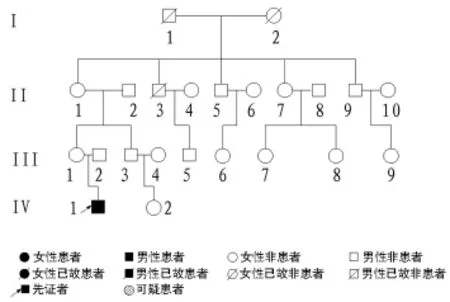
图1 DMD家系示意图
1.2 Dystrophin 基因检测
1.2.1 主要仪器及试剂 台式高速低温离心机;水平及垂直电泳仪系列;PCR仪;MF3-11多功能三维旋混仪;UVP Bio Spectrum R600Imaging System凝胶成像仪;Tag DNA聚合酶,dNTP等。
1.2.2 模板制备与PCR扩增 用快速盐析法提取外周血全基因组DNA,置-20℃冰箱备用。对Dystrophin基因79对引物分别进行扩增,其中第59号外显子引物为:上游 5'CAAAGGGAGTTATCTGTGAGG 3',下游 5'GAATTTGTGAAAGACGGACTG 3',片段长度749 bp,PCR后用于测序和限制性核酸内切酶处理。引物均由武汉擎科新业生物技术有限公司合成。引物PCR反应体系20μL,组份为:灭菌纯水6μL,上下游引物(10μmol/L)各1μL,DNA 模板(50~100 ng/μL)2 μL,2×Taq PCR Green Mix 10 μL。两对引物退火温度均为54.5℃,共36个循环。
1.2.3 DNA直接测序及限制性核酸内切酶分析(RELP) 由武汉擎科新业生物技术有限公司将所需测序样本PCR产物用双脱氧末端终止测序法行DNA序列测定。限制性核酸内切酶处理:内切酶Msp1,切点 TC CGG,20 μL体系中,内切酶(Msp1)0.5 ~2 μL(10 U/μL)、DNA(0.5 ~1 μg/μL)1 μL、10×Buffer Tango 2μL,37℃水浴过夜。处理后,用1.5%琼脂糖水平跑胶,凝胶成像系统照相并保存。
2 结果
2.1 DNA直接测序结果 先证者行Dystrophin基因所有外显子的测序。测序结果用Chromas软件进行BLAST分析,再将测序峰与网上检索(http://www.ncbi.nlm.nih.gov/)结果重新比对。先证者Dystrophin基因第9017位发生G→A的纯合突变(上下为正反向测序图),在网上序列比对后得出,此为错义变异,其所代表的精氨酸的2937位密码子变化为谷氨酰胺(R2937Q),见图2。
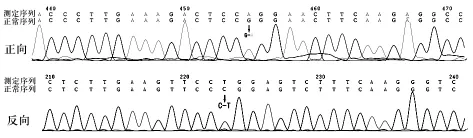
图2 DNA直接测序结果
2.2 RELP结果 先证者9017位位点发生纯和变异,故没有被酶切(Ⅳ1);家系内其他成员和正常对照者9017位位点未发生变异,故被酶切成两段(256 bp和493 bp)。
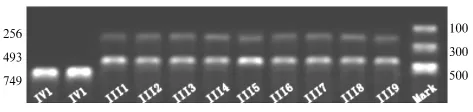
图3 先证者及其他家系成员、健康对照者基因变异位点PRLP结果
3 讨论
DMD是一种遗传性、进行性加重的肌肉疾病,其特征为进行性近端肌肉无力,伴肌纤维的破坏与再生,被结缔组织所取代[5]。DMD患者多预后不良,一般学龄期初诊[6],多因呼吸衰竭或心脏衰竭等并发症而死亡。其遗传特征多符合X染色体隐性遗传,由于女性有两条X染色体,当隐性致病基因在杂合状态时,隐性基因控制的性状或遗传病不显示出来,此为女性表型正常的致病基因携带者,只有当两条X染色体上等位基因都是隐性致病基因纯合子时才表现出来。而男性只有一条X染色体,Y染色体上缺少同源节段,所以只要X染色体上有一个隐性致病基因就发病[7]。
鉴于DMD明显的遗传特征,国内外许多学者对此进行了研究。目前较为肯定的致病基因是Dystrophin基因,其包括79个外显子,全长约为2 500 kb。其mRNA序列长1.4 kb,主要表达于骨骼肌、心肌,少量表达于脑组织[7~12]。Dystrophy 基因按照功能划分可分为四部分:氨基端区域(1~8号外显子)、中央棒状区(9~62号外显子)、富含半胱氨酸区域(63~69号外显子)、羧基端区域(70~79号外显子)。之前已知的突变类型中最为常见的为基因缺失,存在2个缺失热区,一个位于中央区域,是最多见的缺失区域,多包括45~55号外显子,另一个缺失热区位于基因的5'端,多包括2~20号外显子。有报道称,缺失热区中外显子缺失断裂点的分布在亚洲及欧洲人群中存在差异[13~16]。
鉴于人种的差异,本研究检测了一个中国DMD家系Dystrophin基因突变类型。结果发现,先证者出现了一个碱基纯合错义变异,是位于第59号外显子(c.9017G>A),导致代表精氨酸的2937位密码子突变为谷氨酰胺(p.Arg 2937Gln),而对其家系进行检测后,其遗传类型并不符合X染色体隐性遗传规律。目前该突变尚未在其他文献及DMD数据库中报道。进一步对正常对照者进行筛查,初步排除基因多态性的可能,因而推测此为一个碱基突变。由于此突变在此家系先证者中出现,故考虑此位点的碱基突变与家系DMD的致病可能有密切的关联。Dystrophy基因第59号外显子位于中央棒状区,其转录可能会影响Dp427蛋白,后者主要表达于脑、肌肉等组织中。推测Dystrophy基因第59号外显子中的错义突变可能会影响上述蛋白的结构或功能,从而导致一些临床症状的发生。
本研究在我国DMD家系先征者中发现了Dystrophin基因一个新的突变位点,说明我国DMD患者可能存在与国外不同的基因突变位点。其家系遗传可能并不符合X染色体隐性遗传规律,故目前应进一步进行家系研究,并对新发现的突变进行相关细胞生物学表达试验。
[1]Bushby K,Finkel R,Birnkrant DJ,et al.Diagnosis and management of Duchenne muscular dystrophy,part 1:diagnosis,and pharmacological and psychosocial management[J].Lancet Neurol,2010,9(1):77-93.
[2]Muntoni F,Torelli S,Ferlini A.Dystrophin and mutations:one gene,several proteins,multiple phenotypes[J].Lancet Neurol,2003,2(12):731-740.
[3]Prior TW,Bridgeman SJ.Experience and strategy for the molecular testing of Duchenne muscular dystrophy[J].JMol Diagn,2005,7(3):317-326.
[4]Blake DJ,Weir A,Newey SE,et al.Function and genetics of dystrophin and dystrophin-related proteins in muscle[J].Physiol Rev,2002,82(2):291-329.
[5]黎青,李少英,张楚敏,等.MLPA技术用于假性肥大型肌营养不良症产前基因诊断的价值[J].中华妇产科杂志,2013,48(3):161-164.
[6]Bushby KMD,Hill A,Steele JG.Failure of early diagnosis in symptomatic Duchenne muscular dystrophy[J].Lancet,1999,353(9152):557-558.
[7]朱海燕,邬玲仟,梁德生,等.DMD基因突变及突变检测技术研究进展[J].基础医学与临床,2005,25(11):975-981.
[8]Ervasti JM.Dystrophin,its interactions with other proteins,and implications for muscular dystrophy[J].Biochim Biophys Acta,2007,1772(2):108-117.
[9]Chen WJ,Lin QF,Zhang QJ,et al.Molecular analysis of the dystrophin gene in 407 Chinese patients with Duchenne/Becker muscule dystrophy by the combination of multiplex ligation-dependent probe amplification and Sanger sequencing[J].Clin Chim Acta,2013,423:35-38.
[10]王皖骏,朱海燕,朱瑞芳,等.假肥大型肌营养不良家系的基因检测与产前诊断[J].中华医学遗传学,2013,30(1):45-48.
[11]Snow WM,Fry M,Anderson JE.Increased density of dystrophin protein in the versus the vermal mouse cerebellum[J].Cell Mol Neurobiol,2013,33(4):513-520.
[12]Suzuki H,Kameyama T,Ohe K,et al.Nested introns in an intron:evidence of multi-step splicing in a large intron of the human drstrophin pre-mRNA[J].FEBSLett,2013,587(6):555-561.
[13]Lai PS,Takeshima Y,Adachi K,et al.Comparative study on deletions of the dystrophin gene in three Asian populations[J].J Hum Genet,2002,47(10):552-555.
[14] Wang Q,Li-Ling J,Lin C,et al.Characteristics of dystrophin gene mutations among Chinese patients as revealed by multiplex ligation-dependent probe amplification[J].Genet Test Mol Biomarkers,2009,13(1):23-30.
[15]Arauio KP,Bonuccelli G,Duarte CN,et al.Bortezomib(PS-341)treatment decreases in inflammation and partially rescues the expression of the dystrophin-glycoprotein cpmplex in GRMD dogs[J].PLoSOne,2013,8(4):e61367.
[16]Rani AQ,Sasonqko TH,Sulong S,et al.Mutation spectrum of dystrophin gene in malaysian patients with Duchenne/Becker muscular dystrophy[J].JNeuroqenet,2013,27(1-2):11-15.
