CrBn(n=1~6)团簇的结构和磁学性质
2013-06-07关曼曼闫雷兵张文庆河南科技学院河南新乡453003
关曼曼,闫雷兵,张文庆(河南科技学院,河南新乡453003)
CrBn(n=1~6)团簇的结构和磁学性质
关曼曼,闫雷兵,张文庆
(河南科技学院,河南新乡453003)
从第一性原理出发,利用密度泛函理论中广义梯度近似对CrB(nn≤6)团簇的基态结构、电子结构和磁性做了系统研究.结果表明:CrB4团簇比较稳定,CrBn团簇的基态结构可看作是添加Cr原子到Bn基态团簇上,与纯Bn+1团簇很相似,Cr掺杂增强了硼团簇的化学活性.Mulliken布局分析表明CrBn体系的磁矩主要来自局域d电子的贡献,n=1和5时,CrBn磁矩最大.
CrBn团簇;密度泛函理论;结构与稳定性;磁性
近年来,过渡金属掺杂B团簇体系引起了人们广泛的研究兴趣.Deshpande等利用密度泛函理论研究了NinB(n=1~8,12)团簇的结构和磁性[1].孙强等利用原子轨道的线性组合方法对FenB(n≤6)团簇的结构和磁性做了研究[2].另一方面,杨致等利用广义梯度近似对FeBn(n≤6)团簇的结构和磁性进行了探讨[3].雷雪玲等计算了Ni掺杂B[4-5],Yao等研究了Zr掺杂B团簇的结构与磁性,指出掺杂的过渡金属原子对团簇的稳定性及电磁性质有很大影响[6].本文将对Cr掺杂B小团簇的结构、电子和磁学性质进行研究,对进一步了解过渡金属元素掺杂对B团簇物理化学性质的影响及合成新的功能材料都有一定的意义.
1 计算方法
计算采用基于密度泛函理论的Dmol3软件包[7],选用了广义梯度近似GGA(BLYP)方法,在电子结构计算中,采取带极化的双数值原子基组(DNP)进行.在结构优化过程中,力的收敛标准是0.020Hartree/nm,位移收敛标准5.000×10-4nm,能量收敛标准10-5Hartree.自洽场收敛标准为10-6Hartree,使用了DIIS方法,轨道计算中smearing标准为0.002Hartree.采取自旋非限制方法,对所有可能的自旋多重度优化.在结构优化过程中对振动频率进行了计算,得到的稳定构型均无虚频.
2 结果与讨论
2.1 团簇的生长行为
为了与纯B团簇对比,用同样的计算方法优化了Bn+1(n≤6)几何结构,纯Bn+1及CrBn(n≤6)团簇基态结构见图1.对于n≤5,纯Bn及CrBn团簇均采取平面结构做为它们的最低能量结构,CrBn(2≤n<5)团簇的基态结构通过桥接一个Cr原子在纯Bn团簇的B-B边上得到.CrB二聚体(图1-1b,自旋多重度为6)键长为0.214 nm,振动频率为424.500 cm-1.CrB2最低能量结构(图1-2b,自旋多重度为3)是具有C2v对称性的等腰三角形结构.CrB3的基态(图1-3b,自旋多重度为4)是具有Cs对称性近的菱形结构.CrB4、CrB5的基态(图1-4b、1-5b,自旋多重度分别是5和6)均是具有Cs对称性的Z字型结构.B6的基态(图1-5a)是一个五棱锥,具有C5v对称性,另一个平面Z字型结构(图1-5a’),具有C2h对称性,在能量上仅比5a结构高0.006 eV,二者可看成是简并态.CrB5的基态结构也可看作是Cr原子替代Z字型结构B6(图1-5a’)一个B原子得到.B7的基态(图1-6a)是一个具有C2v对称性近平面的六边锥结构,替代B7六边锥锥底边一个B原子优化后得到CrB6的最低能量结构(图1-6b),该结构是一个具有C2v对称性平面结构,自旋多重度为3.
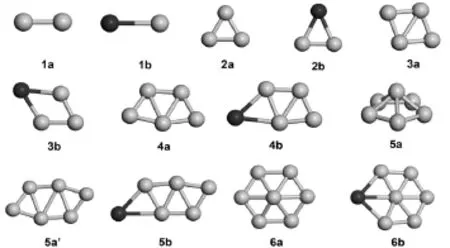
图1 CrBn和Bn+1(n≤6)团簇的基态结构Fig.1 Ground state structureofCrBnand Bn+1(n≤6)clusters
从图1中可以看到,对于CrBn(n≤6)基态团簇,Cr原子替代Bn+1团簇中的一个B,趋向于成为Bn+1团簇的一部分,并且能被看成是在纯Bn+1团簇中的替代杂质,整个团簇增长趋势表明CrBn基态结构与纯Bn+1团簇很相似,在边位置的Cr原子几乎不影响主团簇B的结构.
2.2 基态团簇的相对稳定性
图2中给出了Bn+1和CrBn基态团簇的平均结合能(Eb)随团簇B原子数的变化情况,可以看出,二者的平均结合能均随总原子数的增加而单调增加,与Bn+1团簇相比,相同n值的CrBn团簇的平均结合能低于Bn+1团簇,说明Cr原子掺杂Bn团簇使体系的稳定性减弱.而文献[4]和[6]报道的Ni、Zr原子掺杂使纯B团簇的稳定性增强,说明不同的过渡金属原子掺杂对B团簇稳定性的影响是不同的.
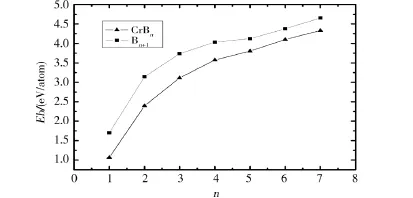
图2 CrBn和Bn+1(n≤6)团簇的平均结合能Fig.2 Binding energies per atom for CrBn and Bn+1(n≤6)clusters
图3 分别给出了CrBn团簇总能的二阶差分(Δ2E)和裂化能ΔE随团簇B原子数变化的情况.裂化能定义如下:ΔE=E(B)+E()-E(CrBn),式中E代表最稳定团簇的总能量.由图3可以看出n=4时,Δ2E和ΔE均为峰值,表明CrB4与邻近的团簇相比具有较高稳定性.
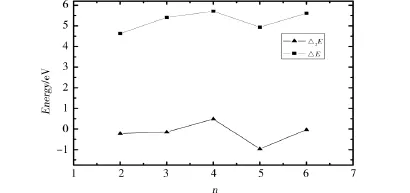
图3 CrBn团簇的裂化能和总能的二阶差分Fig. 3 Fragmentation energy and the second differences of CrBn clusters energies
2.3 基态团簇的电子性质
图4分别给出了CrBn团簇和纯Bn+1团簇的最高占据分子轨道与最低未占据分子轨道(HOMO-LUMO)能隙Eg和垂直电离势VIP随B原子数变化的情况.CrBn团簇的能隙奇偶振荡,n为偶数的CrBn团簇能隙较大,具有较弱的化学活性,稳定性强.与纯硼团簇相比,除n=6外,CrBn团簇的能隙比纯Bn+1团簇都小,说明Cr掺杂增强了硼团簇的化学活性.从图4中还可能看到,CrBn团簇的VIP比纯Bn+1团簇小,说明Cr掺杂使硼团簇易于电离,金属性增强.

图4 CrBn和Bn+1(n≤6)团簇的能隙和垂直电离势Fig. 4 Vertical ionization potentials and HOMO-LUMO gaps for CrBn and Bn+1(n≤6)clustersnand Bn+1(n≤6)clusters
2.4 基态团簇的磁性
Mulliken布局分析的结果见表1.
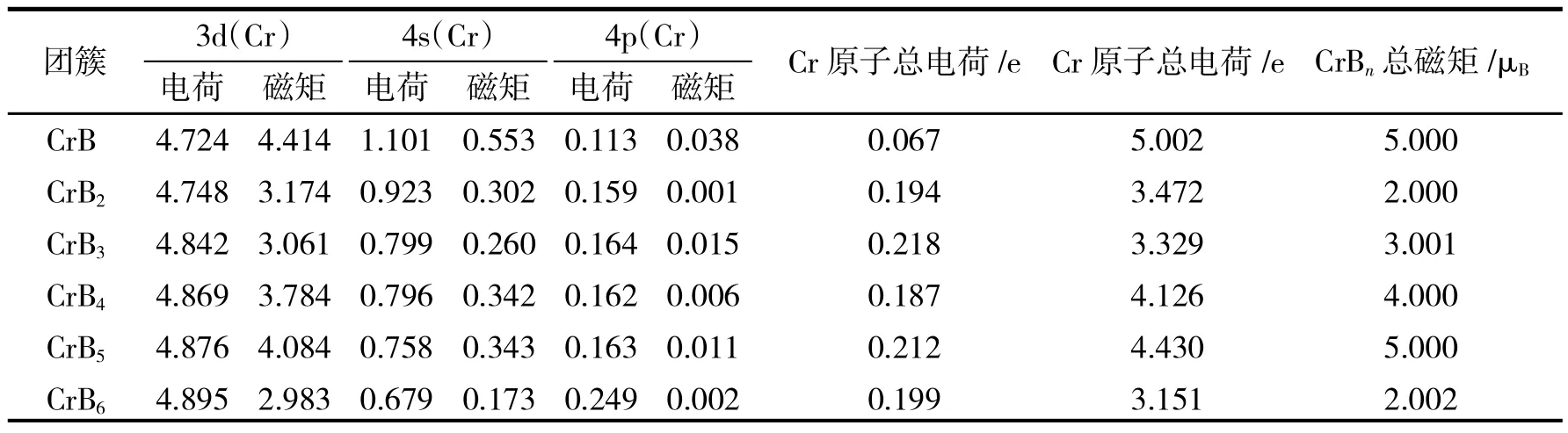
表1 CrBn(n≤6)团簇轨道电子占据数和磁矩Tab. 1 The orbital populations and magnetic moments of CrBn(n≤6)clusters.n(n≤6)clusters.
由表1可知,CrBn团簇的总磁矩范围分别在2.000~5.000μB之间,比较Cr原子磁矩和CrBn团簇的总磁矩可以看出,团簇的总磁矩主要由Cr原子提供.除n=5外,Cr原子的磁矩均大于CrBn团簇的总磁矩,说明B、Cr原子磁矩方向相反,它们之间存在反铁磁耦合.CrB、CrB5团簇的磁矩最大,均为5μB.
从表1中还可以看出,Cr原子3d轨道电子的磁矩在2.983~4.414μB之间,4s和4p轨道电子的磁矩在0.001~0.553μB,可见Cr原子的磁矩主要由其3d轨道电子提供.自由Cr原子(3d54s1)和B原子(2s22p1),n=1除外,CrBn中Cr原子的3d和4s轨道失去电子,4p轨道得到了额外的电子,结果Cr原子的3d和4s轨道向自身的4p轨道转移.另一方面,Cr原子净布居为正值说明CrBn团簇中电荷从Cr原子向B原子转移,是因为B比Cr原子的电负性大,是电子受体的缘故.所以在CrBn团簇中电荷转移主要发生在Cr原子的4s、3d和4p及B原子的2s和2p轨道之间,在Cr原子中存在着sd-p轨道杂化,B原子中存在着s-p轨道杂化.
3 小结
利用密度泛函理论中的广义梯度近似对Bn+1及CrBn(n≤6)小团簇进行了研究,得到如下结论:①CrBn与纯Bn+1团簇的基态结构很相似,CrBn团簇的基态结构可看作是Cr原子替代Bn+1基态团簇一个B原子得到.②通过对平均结合能、团簇的二阶能量差分和裂化能的分析表明,CrB4与邻近的团簇相比具有较高稳定性.与Bn+1团簇相比,相同n值的CrBn团簇的稳定性低于Bn+1团簇.③CrBn团簇的VIP比纯Bn+1团簇小,易于电离,除n=6外,CrBn团簇的能隙比纯Bn+1团簇都小,说明Cr掺杂增强了硼团簇的化学活性.④CrBn团簇总磁矩主要由Cr原子提供,主要来自局域d电子的贡献,除CrB5外,CrBn团簇中Cr和B原子自旋极化方向取向相反,存在反铁磁耦合.
[1]DeshpandeM,Kanhere DG,Pandey R.Structures,energetics,andmagnetic properties ofNinB clusterswith n=1~8,12[J].Phys Rev A,2005,71(6):063202.
[2]孙强,龚新高,郑庆祺,等.Fe-B团簇的结构与磁性的第一性原理研究[J].物理学报,1996,45(7):1146-1152.
[3]杨致,闫玉丽,赵文杰,等.FeBN(N≤6)团簇的结构和磁性[J].物理学报,2007,56(5):2590-2595.
[4]雷雪玲,王清林,闫玉丽,等.利用密度泛函理论研究BnNi(n≤5)小团簇的结构和磁性[J].物理学报,2007,56(8):4484-4490.
[5]雷雪玲,祝恒江,葛桂贤,等.密度泛函理论研究BnNi(n=6~12)团簇的结构和磁性[J].物理学报,2008,59(9):5491-5499.
[6]Yao JG,Wang XW,Wang YX.A theoreticalstudyon structuraland electronic propertiesofZr-doped B clusters:ZrBn(n=1~12)[J]. ChemicalPhysics,2008,351(1):1-6.
[7]Delley B.An all-electron numericalmethod for solving the local density functional for polyatomic molecules[J].JChem Phys, 1990,92(1):508-517.
(责任编辑:卢奇)
Study on structures and magnetism properties of CrBn(n=1~6)clusters
Guan Manman,Yan leibing,ZhangWenqing
(Henan InstituteofScienceand Technology,Xinxiang453003,China)
The ground-state structural,electronic and magnetic properties of CrBn(n≤6)clusters have been investigated by using of the generalized gradient approximation density functional theory.The results indicate that CrB4is themost stable of the clusters.Also,the ground state structures of CrBnclusters can be obtained by directly adding Cr atom to Bn clusters,resembling the Bn+1species.And dopant of the Cr atom enhances the chemical reactivity of the clusters.By Mulliken population analysis,the result shows that themagnetic moment of CrBn clusters mainly comes from the localized d electron,Themagnetic moment of the CrBn clusters(n=1 and 5)is the biggest among all of the CrBn clusters.
CrBn clusters;density functional theory;structure and stability;magnetism
O641
A
1008-7516(2013)02-0060-04
10.3969/j.issn.1008-7516.2013.02.015
2013-03-03
河南省教育厅科学技术研究重点项目(13B140014);河南省教育厅自然科学研究计划项目(2011B140008);河南科技学院大学生课外科技活动创新基金(20110315)
关曼曼(1989-),女,河南夏邑人.主要从事纳米磁性颗粒研究.
张文庆(1972-),男,河南新乡人,硕士,副教授.主要从事计算凝聚态物理研究.
