加味皮康颗粒的质量标准研究
2013-06-07王晓飞葛海生
于 玲 王晓飞 葛海生
武警部队药品仪器检验所,北京 102613
加味皮康颗粒的质量标准研究
于 玲 王晓飞 葛海生
武警部队药品仪器检验所,北京 102613
目的:建立加味皮康颗粒的质量标准。方法:采用薄层色谱法对方中的桑白皮、白鲜皮、当归进行定性鉴别,采用高效液相色谱法对芍药苷进行含量测定。结果:TLC斑点清晰,分离度好,专属性强,阴性对照无干扰;芍药苷进样量在0.01394~0.5576μg范围内线性良好(r=0.9999),平均回收率:98.45%,RSD:1.33%(n=6)。结论:所建标准能有效控制加味皮康颗粒的质量。
加味皮康颗粒;质量标准;薄层色谱法;高效液相色谱法;芍药苷
加味皮康颗粒是由牡丹皮、桑白皮、白鲜皮、当归、陈皮、五加皮、大腹皮、地骨皮等多味中草药组成,具有清热凉血,祛风止痒,活血消斑的功效,临床用于玫瑰糠疹、湿疹的治疗,为保证和控制该制剂的质量,本文建立了白鲜皮、桑白皮、当归的薄层色谱(TLC)定性鉴别,采用高效液相色谱(HPLC)测定制剂中芍药苷的含量,方法简单,专属性强、重复性好,获得了较满意的结果。
1 仪器与试药
1.1 仪器 SHIMADZU LC-2010HTC高效液相色谱仪,METTLER TOLEDO AL204电子天平。
1.2 试药 白鲜皮对照药材(批号:120978-201105,购自中国食品药品检定研究院)、桑白皮对照药材(批号:12114-201006,购自中国食品药品检定研究院)、当归对照药材(批号:120927-201014,购自中国食品药品检定研究院),芍药苷对照品(批号:110736-200526,购自中国食品药品检定研究院);加味皮康颗粒(批号:20130301、20130302、20130303,为武警部队药品仪器检验所自制);硅胶G薄层板(青岛海洋化工厂分厂);乙腈、甲醇为色谱纯,其他试剂为分析纯。
2 定性鉴别:
2.1 桑白皮的TLC鉴别[1]取本品约4.5g,加饱和碳酸钠溶液30ml,超声20分钟,加稀盐酸调pH值至1~2,静置30分钟,用乙酸乙酯提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液Ⅰ。另取桑白皮对照药材2g,按供试品溶液Ⅰ制备的方法制成对照药材溶液Ⅰ;另取不加桑白皮药材的阴性样品,按供试品溶液Ⅰ制备的方法制成阴性对照溶液Ⅰ。按薄层色谱法(《中国药典》一部2010年版附录ⅥB)试验,吸取上述溶液供试品溶液Ⅰ5μl、对照药材溶液Ⅰ10μl,阴性对照溶液Ⅰ5μl,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-乙酸乙酯(6:4)为展开剂,展开、取出、晾干。置紫外灯(365nm)下检视,结果供试品溶液色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,阴性对照溶液在相应位置上无干扰(见图1)。

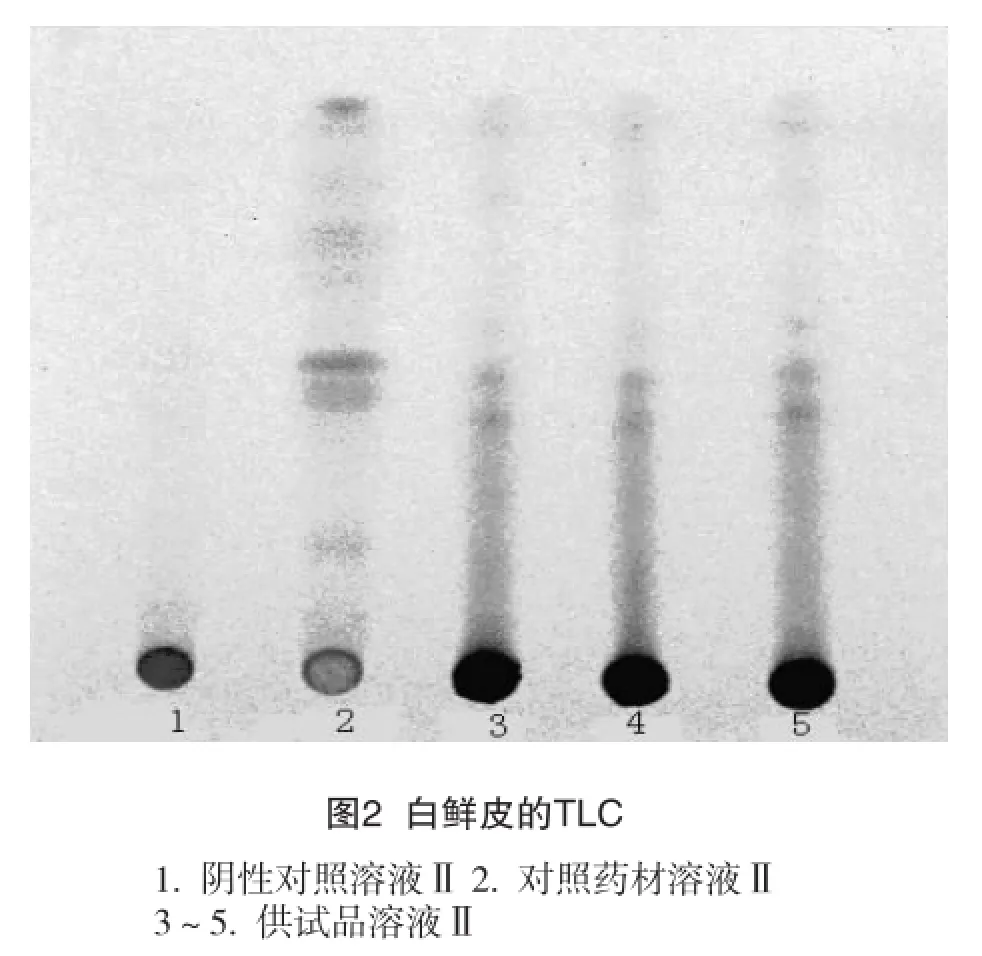
2.2 白鲜皮的TLC鉴别[2]取本品约2.3g,加甲醇30m l,超声20分钟,滤过,滤液加中性氧化铝(100~200目)1.5g,振摇2min,滤过,滤液置水浴上蒸干,残渣加甲醇1m l使溶解,作为供试品溶液Ⅱ。另取白鲜皮对照药材1g,加水40ml,煮沸并保持微沸30min,脱脂棉滤过,滤液置水浴上蒸干,残渣加甲醇2m l使溶解,作为对照药材溶液Ⅱ。另取不加白鲜皮药材的阴性样品,按供试品溶液Ⅱ制备的方法制成阴性对照溶液Ⅱ。按薄层色谱法(《中国药典》一部2010年版附录ⅥB)试验,吸取上述溶液各5μl,分别点于同一硅胶G薄层板上,以环己烷-丙酮-浓氨水(20:20:1)的上层溶液为展开剂,展开、取出、晾干。喷以10%硫酸乙醇溶液,105℃加热至斑点清晰。结果供试品溶液色谱中,在与对照药材色谱相应的位置上,分别显相同颜色的斑点,阴性对照溶液在相应位置上无干扰(见图2)。
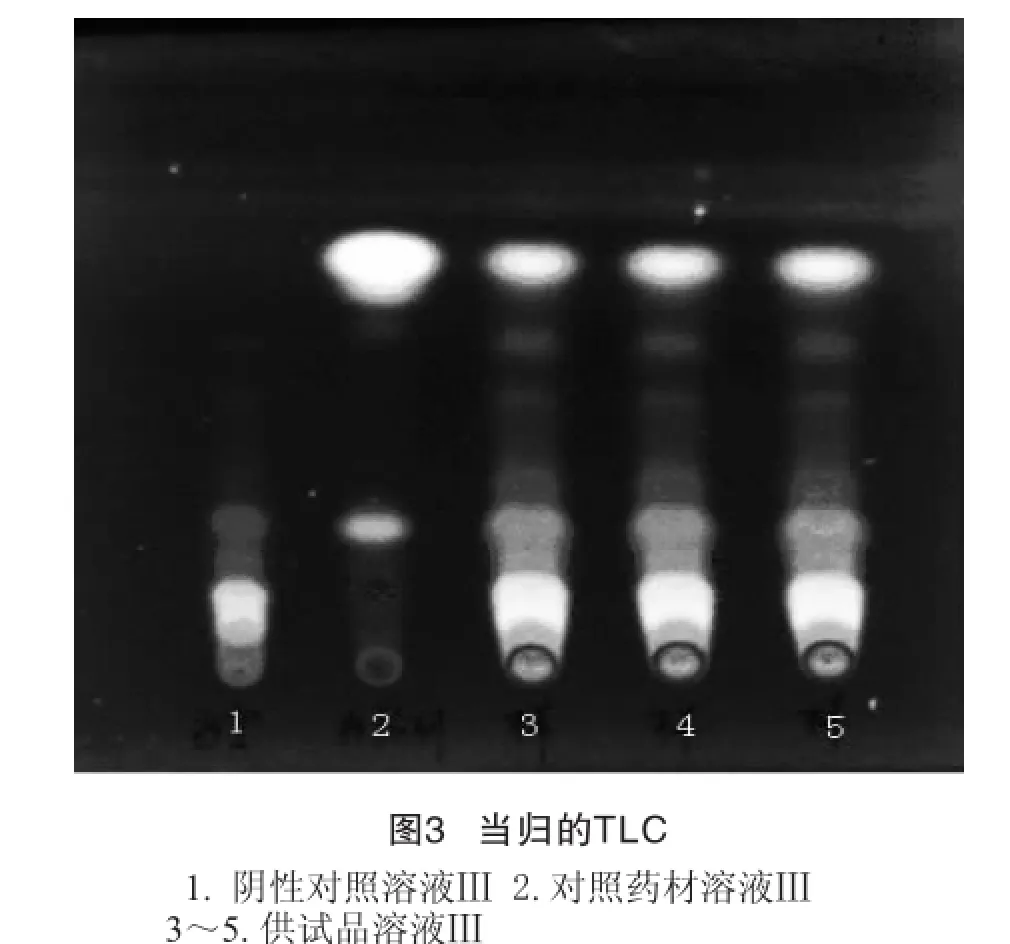
2.3 当归的TLC鉴别[3]取本品2.3g,加1%碳酸氢钠溶液20m l,超声处理10分钟,离心,取上清液用稀盐酸调节pH值至2~3,用乙醚振摇提取2次,每次20m l,合并乙醚液,挥发,残渣加甲醇1m l使溶解,作为供试品溶液Ⅲ。另取当归对照药材0.5g,按供试品溶液Ⅲ制备的方法制成对照药材溶液Ⅲ;另取不加当归药材的阴性样品,按供试品溶液Ⅲ制备的方法制成阴性对照溶液Ⅲ。按薄层色谱法(《中国药典》一部2010年版附录ⅥB)试验,吸取上述溶液各10μl,分别点于同一硅胶G薄层板上,以环己烷-二氯甲烷-乙酸乙酯-甲酸(4:1:1:0.1)为展开剂,展开、取出、晾干。置紫外灯(365nm)下检视,结果供试品溶液色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,阴性对照溶液在相应位置上无干扰(见图3)。
3 芍药苷的含量测定[4]
3.1 色谱条件 色谱柱:SHISEIDO,ODS分析柱(4.6mm ×250mm,5?m);流动相:乙腈-0.1%磷酸溶液(14:86),柱温:30℃,检测波长:230nm,流速:1.0m l·min-1,在上述色谱条件下,芍药苷和供试品中其它组分可达到基线分离,芍药苷与其相邻色谱峰的分离度大于1.5,且阴性对照液无干扰(附图);理论塔板数按芍药苷峰计大于2000。
3.2 对照品溶液的制备 精密称取芍药苷对照品约6.97mg,置于10m l量瓶中,加甲醇适量溶解后,稀释至刻度,摇匀制得贮备液。精密吸取贮备液1m l,置50m l量瓶中加甲醇稀释至刻度,作为对照品溶液。
3.3 供试品溶液的制备 取本品约1g,精密称定,置于锥形瓶中,精密加入甲醇25m l,称定重量,超声处理(功率300W,频率45kHz)15分钟,放冷,再称定重量,加甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
3.4 阴性对照溶液的制备 按处方配制不含牡丹皮的样品颗粒,并按“3.3”项下方法对样品进行制备,制成阴性样品溶液。
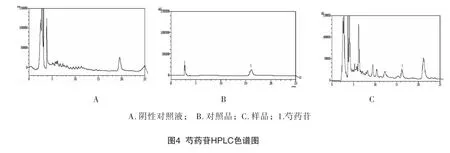
3.5 方法专属性试验精密吸取芍药苷阴性对照溶液、对照品溶液、供试品溶液各10μl,分别注入色谱仪,按“3.1”项下色谱条件进行测定,记录图谱,见图4。在B、C色谱图相应的位置上具有相同保留时间色谱峰,而A图在相应位置上无干扰峰。
3.6 线性关系考察精密量取对照品溶液(浓度为:13.94μg·m l-1)各1,5,10,15,20,25,30,35,40μl注入色谱仪中,按“3.1”项下色谱条件测定芍药苷峰面积(A),作为纵坐标,同时以芍药苷进样量(μg)为横坐标,得回归方程为Y=766+1254293X,r=0.9999。以上数据表明芍药苷进样量在0.01394μg~0.5576μg范围内线性良好。
3.7 精密度试验 精密量取3.6中浓度为13.94μgμml-1的溶液,按3.1项下色谱条件重复进样6次,每次10μl,计算得芍药苷的峰面积RSD为0.86%(n=6),表明仪器的精密度良好。
3.8 稳定性试验将3.3项下的溶液于室温放置,分别在放置0,2,4,6,8,10,12h时进行分别进行测定,计算得芍药苷峰面积RSD为1.45%(n=7),表明供试品在12h内基本稳定。
3.9 重复性试验 取同一批号的样品6份,按样品溶液制备方法制备及测试条件测定,结果芍药苷平均含量的RSD=1.31%(n=6),表明本法重复性良好。
3.10 回收率试验 采用加样回收法,取已知芍药苷含量的同一批号的本品(批号:20130302,含量为0.2315 mg· g-1)约1g,共6份,精密称定,置于锥形瓶中,精密加入芍药苷对照品溶液(浓度为0.2323mg·ml-1)各1ml,再精密加入甲醇25ml,按3.3中项下方法制成供试品,分别吸取供试品溶液10μl,注入HPLC仪,测定芍药苷含量,计算加样回收率,结果见表1。
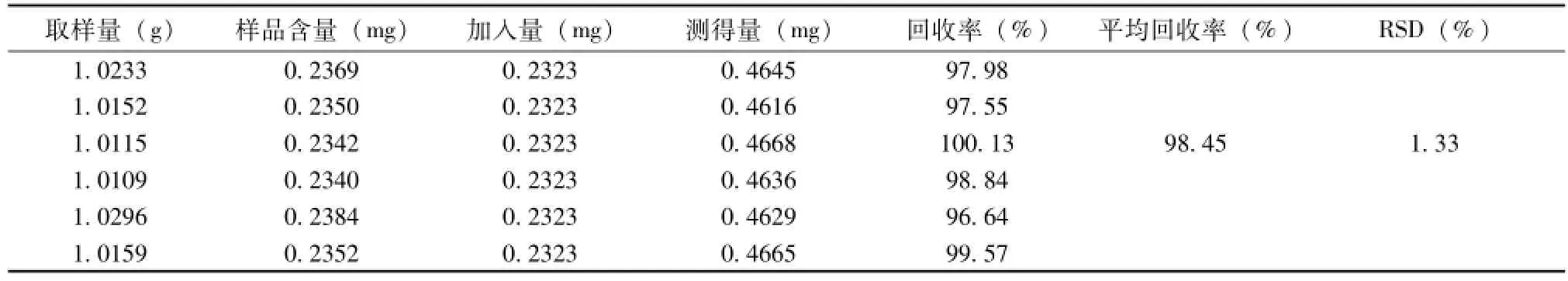
表1 加样回收率试验结果(n=6)
3.11 样品的测定 取本品3批,按“3.3”项下制成供试品溶液,进样10μl分别测定。按外标法计算芍药苷的含量,结果见表2。
表2 样品含量测定结果(n=3)
4 讨论
4.1 在2.1桑白皮的TLC鉴别中,曾采用样品溶液点于聚酰胺薄膜上,展开剂用醋酸,进行试验,分离结果不满意。
4.2 在2.2白鲜皮的TLC鉴别中,曾采用样品溶液超声后,滤过,滤液未加中性氧化铝(100~200目)振摇,滤过,滤液直接蒸干,残渣加甲醇使溶解,制成供试品后进行试验,分离结果不满意。
4.3 本试验对方中的主要成分桑白皮、白鲜皮、当归采用TLC进行定性鉴别,色谱斑点清晰,分离效果好,专属性强,可用于该制剂的定性鉴别。
4.4 在定量试验中,取同一批号的样品,采用甲醇、乙醇、稀乙醇分别考察了超声处理15min的考察方法,结果表明,以甲醇为溶剂,超声处理15min,方法简便,提取充分,方法最佳。
4.5 试验中,比较了参考文献4、5所述的流动相、柱温,以乙腈-0.1%磷酸溶液(14:86),柱温:30℃,制剂中芍药苷能与其他组分的色谱峰很好分离,理论塔板数按芍药苷峰计大于2000,且方法简便、实用,结果准确,重现性好,可有效的控制加味皮康颗粒的质量。
[1]贺云杰,赵晨,李妍,等.参附强心丸中人参、大黄、桑白皮的薄层鉴别鉴别及乌头碱限度检查[J].世界科学技术(中医药现代化),2009,03:468.
[2]王春桃,符洪.白山片的薄层色谱鉴别方法研究[J].中国热带医学,2008,8(6):1036.
[3]国家药典委员会编.中华人民共和国药典(一部)[S].2010年版.北京:化学工业出版社,2010:124.
[4]国家药典委员会编.中华人民共和国药典(一部)[S].2010年版.北京:化学工业出版社,2010:97.
[5]王晓飞,葛海生,于玲,等.柴辛鼻敏康颗粒的质量标准研究[J].中国药房,2011,22(19):1800.
Quality Standard of jiawei pikang Granules
Yu Ling,Wang Xiao-fei,Ge Hai-sheng(Institute for Drug Control of the Chinese People's Armed Police Forces,Beijing102613,China)
Objective:To establish the Quality Standard of jiawei pikang Granules.Methods:TLC was adopted to identify Cortex Mori,Radix Ampelopsis and Radix Angelicae sinensis.HPLCmethodwasused to test the contentof paeoniflorin.Results:TLC spots were clear,well-separated and specific without interference from negative control;The linear range of paeoniflorin was0.01394~0.5576?g(r=0.9999)with an average recovery of98.45%,The RSD is 1.33%(n=6).Conclusion:Establish Standard is suitable for the quality control of jiawei pikang Granules.
jiawei pikang granules;quality standard;TLC;HPLC;paeoniflorin
R286
A
1007-8517(2013)23-0014-03
2013.10.13)
于玲,副主任药师,本科,研究方向为药品检验,E-mail:yuling.y@163.com
