罗格列酮对大鼠脑缺血再灌注损伤后IL-18 表达的影响
2013-03-11张宁,刘元
张 宁,刘 元
近年来,探讨炎症反应在缺血性脑梗死中的损伤机制成为研究的热点,抗炎治疗被认为是改善脑梗死病情,降低脑卒中患者致残致死率的有前景的治疗方法之一。过氧化物酶体增殖激活型受体γ(peroxisome proliferator-activated receptorγ,PPARγ)属于Ⅱ型核受体超家族成员,受配体激活后能调控多种基因的转录和表达,除降低血糖水平外,尚可改善动脉粥样硬化、纠正血脂紊乱、抗脏器纤维化等,新近[1~3]研究发现PPARγ 选择性激动剂噻唑烷二酮类药物(TZDs)参与多脏器缺血再灌注后炎症反应过程,但其机制尚未清楚。据此,本实验采用TZDs 类药物马来酸罗格列酮(RSG)对大鼠缺血再灌注后进行药物干预,观察各组大鼠脑梗死体积及炎症因子白介素-18(IL-18)表达变化,试图为脑卒中抗炎治疗提供新的理论依据。
1 材料与方法
1.1 材料
1.1.1 实验动物及分组 250~280g 清洁级健康雄性成年SD 大鼠60 只(湘雅医学院动物实验中心提供),随机分为5 组:假手术组(n=12),缺血再灌注模型组(n=12),罗格列酮0.5mg/kg、2mg/kg、5mg/kg 大中小3 种剂量干预组(3 组各n=12)。Longa 线栓法制备大脑中动脉栓塞模型(MCAO)。罗格列酮干预组于缺血即刻及缺血2h 后经插入胃管分别灌入马来酸罗格列酮0.5mg/kg、2mg/kg、5mg/kg 大中小3 种剂量。假手术组和模型组以相同体积的生理盐水灌胃,在缺血2h 再灌注24h 后处死大鼠。
1.1.2 主要实验药品 罗格列酮(rosiglitazone)购自葛兰素史克公司,IL-18 抗体购买自美国Millipore 公司,PV6001 二抗试剂盒、DAB 显色试剂盒,由北京中杉金桥生物公司提供,其余试剂由湘雅医院神经内科实验室提供。
1.2 方法
1.2.1 脑缺血再灌注动物模型制备及评分参考Longa 的线栓法制备局灶性大脑中动脉脑缺血模型。主要步骤包括:10%水合氯醛(0.35ml/kg)腹腔注射麻醉大鼠,分离颈总动脉、颈外动脉后分别予以结扎,颈总动脉远端作血管切口,沿颈总和颈内动脉缓慢插入头端钝化的尼龙栓线约18±5mm 阻塞大脑中动脉入口,缺血2h 后拔出栓线进行24h 再灌注。假手术组插线深度为8~10mm,其余同模型制备。根据Longa 的5 级标准评分法对大鼠进行评分并记录,其中1~3 分为造模成功。没有神经功能缺损、有呼吸困难、提前死亡及处死时发现有蛛网膜下腔出血的动物均排除。手术中出血过多的动物也弃去。对于实验中死亡或者剔除的大鼠严格按实验条件统一补充。将制作成功的模型麻醉后4%多聚甲醛进行灌注,取自额极起4~11mm 之间的脑组织4%多聚甲醛固定过夜,依次脱水、透明、浸蜡、包埋。将包埋好的石蜡组织块修材,自大脑向后以冠状切面连续切片,每片厚5μm,收集含缺血区的脑片贴片、烤片。
1.2.2 IL-18 免疫组化染色检测 用兔抗大鼠IL-18 多克隆抗体进行免疫组织化学染色,每只实验动物每个指标取两张切片。(1)石蜡切片脱蜡:石蜡切片置60 ℃烤箱烤片15h 以上→从烤箱中拿出立即放入二甲苯Ⅰ10min→二甲苯Ⅱ10min→无水乙醇2min→95%乙醇2min→80%乙醇→70%乙醇→自来水洗。(2)3%H2O2阻断20min(以消除内源性过氧化物酶的活性,降低背景);PBS 洗3 次,每次3min。(3)抗原修复(柠檬酸pH 6.0 修复10min);PBS 洗3 次,每次3min。(4)滴加5%正常山羊血清封闭,37℃,10~20min(消除背景非特异性着色)。(5)吸干组织周围多余的血清,不洗,滴加第一抗体,37℃孵育30min 再4℃冰箱过夜;PBS 洗3 次,每次3min。(6)擦干组织周围PBS,滴加生物素标记的第二抗体,37℃孵育20min,PBS 洗3 次,每次3min。(7)擦干组织周围PBS,滴加链霉菌抗生物素-过氧化物酶溶液,37℃孵育20min。(8)PBS洗3 次,每次3min,蒸馏水洗2 次,DAB 显色(DAB必须现用现配),镜下控制显色程度,显色约5min,自来水冲洗,苏木素复染胞核30s,烤干,中性树胶封片。在生物学显微镜下观察脑缺血侧皮质IL-18阳性细胞,在40×10 倍显微镜下随机选取10 个视野,图像分析软件(Image-proplus-6,美国)分析计算每张切片中所选中区域平均光密度值(AOD)。
1.2.3 病理学观察 石蜡切片HE 染色后光镜下观察脑组织缺血半暗带神经元细胞病理学改变及周围炎性细胞浸润。
1.2.4 TTC 染色法测定脑梗死体积 每组取6 只麻醉处死后取脑放于零下20℃冰箱中冷冻30min 后,切成1.2mm 厚的脑片,迅速放于2%红四氮唑(TTC)磷酸缓冲液中,避光37℃恒温孵育30min。正常组织染为红色,梗死组织为白色。10%的多聚甲醛溶液中固定24h 后拍照,应用病理图形分析仪测量脑梗死体积。根据公式V=t(A1+...An)-(A1+An)t/2 算出脑梗死体积和所染部分体积(其中t 为切片厚度,A 为梗死面积)。
1.3 统计学方法
2 结果
2.1 神经缺陷评分
动物麻醉后约2~3 h 清醒,假手术组动物清醒后无明显神经系统功能缺损的症状和体征。其他各组动物于清醒后出现反应欠佳,活动量及进食明显减少,左侧前肢无力,提起尾时左前肢屈曲不能伸直,行走时左侧偏斜或旋转跌倒现象。与缺血再灌注模型组比较,大中剂量ROSI 治疗组大鼠神经功能缺损评分均明显降低,差异统计学意义(P<0.05)(见表1)。
2.2 罗格列酮对大鼠脑梗死体积的影响
缺血再灌注后,假手术组大鼠脑组织未见梗死灶,模型组大鼠右侧额顶叶和纹状体可见较明显的苍白梗死灶。与假手术组相比,大中剂量组均可降低脑梗死体积P<0.05,有统计学意义(见表2)。
2.3 脑缺血再灌注后缺血半暗带IL-18 表达
IL-18 在神经元细胞及胶质细胞均有表达,光镜下可见棕黄色颗粒沉积于细胞胞浆。假手术组检测到极少量IL-18 阳性表达细胞。与假手术组相比,模型组及小剂量干预组阳性蛋白表达数量明显增加,有统计学差异(P<0.01);与模型组相比,大中剂量干预组病灶处阳性表达数量明显减少,有统计学差异(P<0.01)(见表3、图1)。
2.4 病理组织学观察
光学显微镜下观察到假手术组脑组织HE 染色显示结构正常。模型组表现出典型脑梗死改变:梗死区域神经元细胞数量较少,细胞核破裂分解、固缩坏死,细胞形态肿胀不规则,伴有白细胞浸润。与模型组相比,ROSI 大中剂量治疗组坏死灶相对较小,高倍镜下可以观察到肿胀坏死的细胞,但细胞减少程度较轻,坏死灶边缘炎性细胞浸润较少。
表1 各组大鼠神经功能评分的比较(±s)
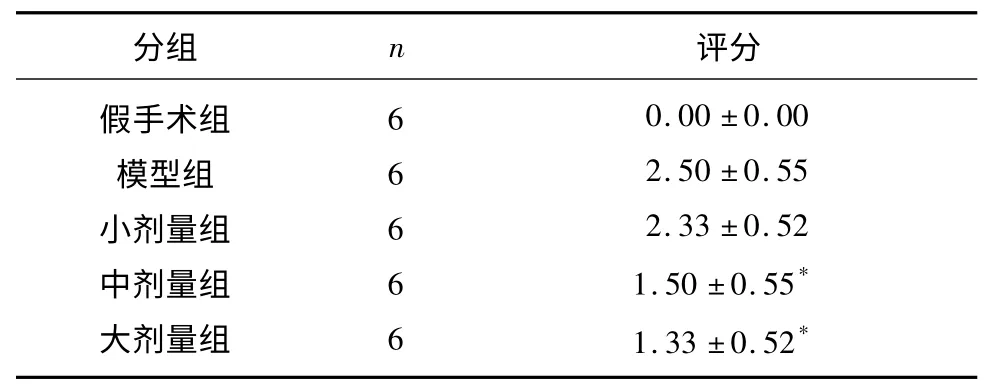
表1 各组大鼠神经功能评分的比较(±s)
与模型组比较* P<0.05
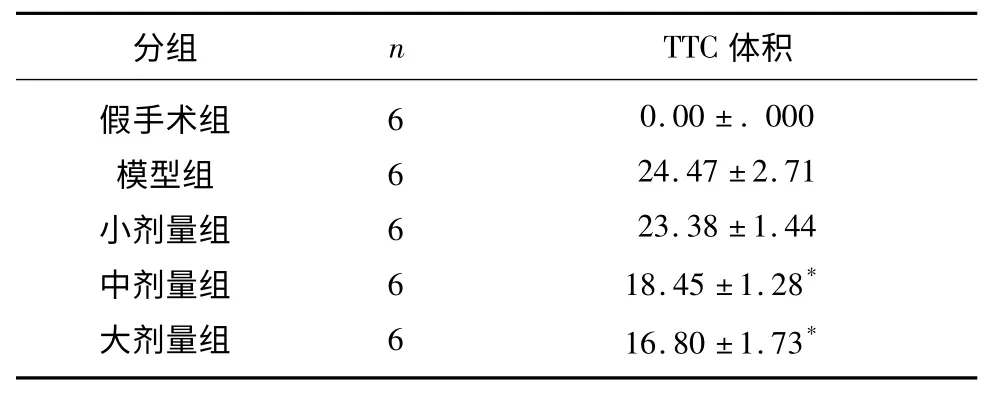
表2 各组大鼠缺血再灌注后脑梗死体积(mm3)
表3 各组大鼠缺血侧顶叶皮质IL-18 表达的比较(±s)
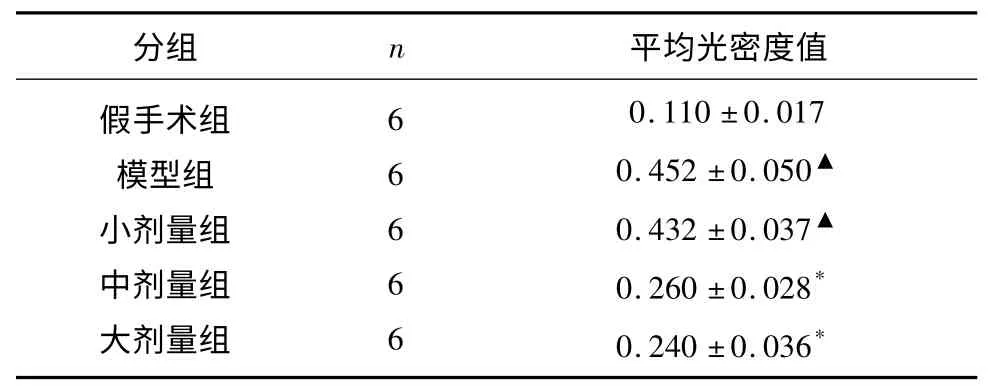
表3 各组大鼠缺血侧顶叶皮质IL-18 表达的比较(±s)
与假手术组比较▲P<0.05;与模型组比较* P<0.05
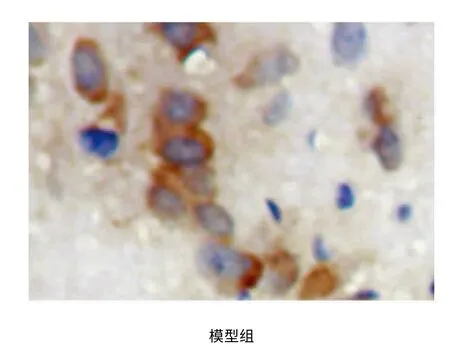
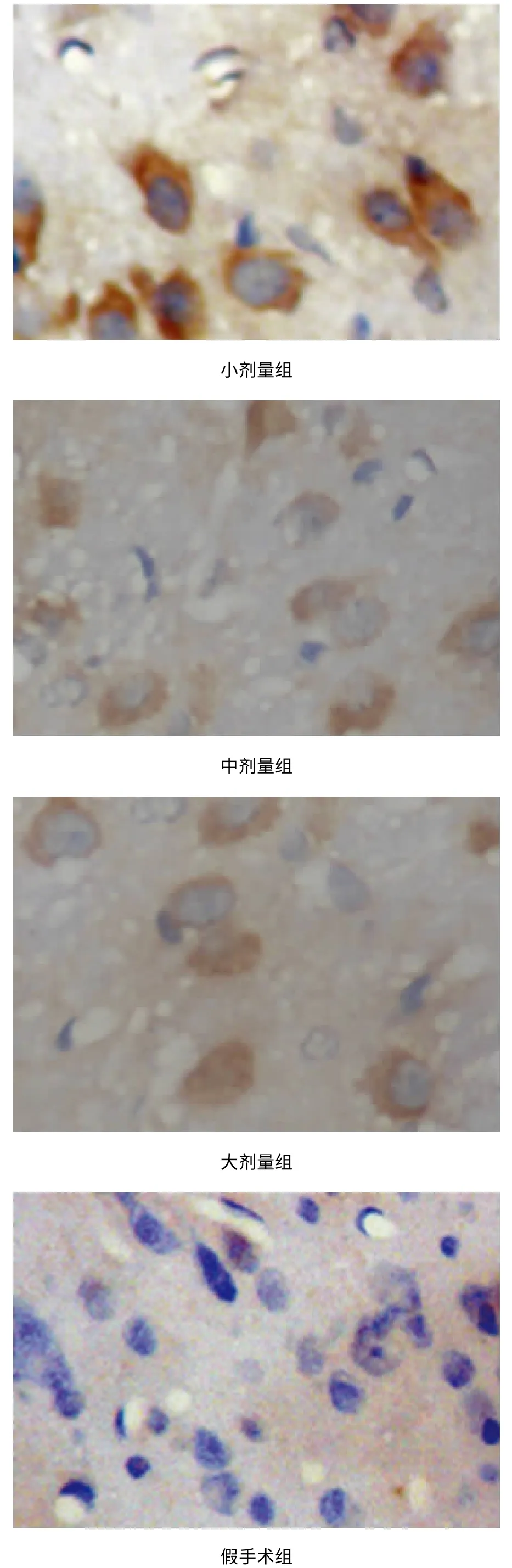
图1 各组大鼠缺血侧顶叶皮质IL-18 表达(IL-18×400)
3 讨论
缺血再灌注引起的脑损伤机制包括多种因素,十分复杂,目前认为其病理生理机制包括缺血后的代谢异常、能量耗竭与酸中毒、兴奋性氨基酸毒性、细胞内钙离子超载、炎症细胞因子损害、氧自由基损害、即早基因、热休克蛋白基因表达以及缺血后迟发性神经细胞凋亡等。其中,缺血再灌注后脑组织内过度的炎症反应是造成再灌注损伤的重要机制之一,而表达增加的炎症因子更被认为是脑缺血再灌注损伤发病机制中的关键介质[4]。因此有效阻断炎症反应是治疗缺血性脑血管疾病的一个重要策略。
炎症反应在脑缺血再灌注引起的继发性损害中起着重要作用.再灌注时各种炎性因子激活炎性细胞,同时参与了神经细胞的凋亡和坏死,在炎症级联反应中起关键作用。IL-18 是近年来发现的具有多种生物学特性的细胞炎性因子。属于IL-1 细胞因子家族成员,主要由单核巨噬细胞系统的细胞分泌,IL-18 具有多种生物学功能,能抑制保护因子IL-4和IL-10 的产生。上调细胞间黏附分子(ICAM)的表达,促进炎性细胞的粘附和聚集,参与神经元细胞损伤。
还能促进致炎因子的释放,特别是具有诱导产生IFN-γ 的强大功能,增强Th1 型细胞反应。此外,IL-18 还可以刺激T 细胞产生IL-2、GM-SF、和TNFα。外周血单核细胞在IL-18 刺激24h 后,可检测到有IL-1 和IL-8 的产生,与Th1 型细胞反应密切相关。有研究显示,IL-18 与心脑血管疾病的发生发展及预后密切相关,参与了肾脏、胃肠道缺血再灌注损伤[5,6]。国内也有研究表明急性脑梗死患者血清IL-18 水平升高;血清IL-18 水平与脑梗死体积及临床神经功能缺损程度密切相关[7]。本研究证实,模型组缺血区脑组织IL-18 表达明显增加,且坏死处白细胞浸润明显增多,表明炎症反应明显参与脑梗死损伤的病理生理过程,进一步印证了IL-18 及其介导的炎性细胞浸润和炎症反应参与缺血/再灌注脑损伤过程。
作为核转录因子,PPARγ 主要通过调节细胞中目的基因转录而发挥其功能。罗格列酮作为PPARγ 的特异性激动剂,其抗炎作用逐渐引起了人们的重视。动物缺血实验显示,PPARγ 经配体激活后可以抑制缺血导致的TNF-α、IL-1β、6、8 等细胞因子过表达,减轻白细胞在缺血区的浸润,降低炎性细胞的毒性作用[8]。国外有研究表明,缺血再灌注后,ROSI 激活PPARγ 可以抑制IL-、TNF-α、IL-6 等诱导的IL-18 表达增加,还可以通过抑制caspase-1阻断IL-18 的激活。此外,IL-18 诱导产生大量的IFN-γ 是其主要生物学功能之一,PPARγ 激活后可以降低IFN-γ 的生物功能,使IFN-γ 激活的单核/巨噬细胞、淋巴细胞仍表现出静止期的功能,并抑制iNO2mRNA、基质金属蛋白酶等炎性介质mRNA 的表达[9,10]我们实验结果也表明,与模型组比较,PPARγ 选择性激动剂罗格列酮2mg/kg、5mg/kg 可明显改善神经缺陷评分、梗死处组织病理学损伤,明显缩小缺血再灌注后脑梗死体积,表明罗格列酮对再灌注后神经损伤具有保护作用,与文献报道的其它噻唑烷二酮类药物效应相似[11]。应用罗格列酮可明显降低炎症分子IL-18 表达和炎性细胞浸润,在大中剂量组表现更显著,而小剂量组未见明显差异,这表明罗格列酮对缺血再灌注脑损伤的保护作用可能与其抑制IL-18 的抗炎效应有关,且可能具有剂量依赖性。
罗格列酮对缺血再灌注损伤的保护作用已引起越来越多的重视,我们的实验显示了罗格列酮能够减少脑梗死面积,改善预后,且其保护作用与抑制炎性介质作用,减少炎性细胞的浸润有关。需要指出的是,缺血再灌注损伤是一个十分复杂的病理生理过程,其机制往往是多方面的,因此,罗格列酮的保护作用可能与其他如抗凋亡、改善缺血后能量代谢、抗自由基、拮抗血小板活化因子、抗兴奋性毒性等药理作用相关联,有关罗格列酮抗脑缺血再灌注损伤的确切分子作用机制正在进一步研究中。
[1]Nakajima A,Wada K,Mik H,et al.Endogenous PPARγmediates antiinflammatory activity in marine ischemia perfusion injury[J].Gastroenterology,2001,120:460-469.
[2]Okada M,Yan SF,Pinsky DJ.PPARγ activation suppresses ischemic Induction of Egrl and its inflammatory gene targets targets[J].FASEBJ,2002,16:1861-1868.
[3]Wayman NS,Attori Y,McDonald M,et al.Ligands of the PPAγreduce myocardial infarct size[J].FASEBJ,2002,16:1027-1040.
[4]Terao S,Yilmaz G,Stokes KY,et al.Inflammatory and injury responses to ischemic stroke in obese mice[J].Stroke,2008,39(3):943-950.
[5]Wu H,Craft ML,Wang P,et al.IL-18 contributes to renal damage after ischemia-reperfusion[J].J Am Soc Nephrol,2008,19(12):2331-2341.
[6]Yang YJ,Shen Y,Chen SH,et al.Role of interleukin 18 in acute lung inflammation induced by gut ischemia reperfusion[J].World J Gastroenterol,2005,11(29):4524-4529.
[7]魏 芳,汤永红,刘卓然.急性脑梗死患者血清白细胞介素-18水平及临床意义[J].南华大学学报医学版,2007,35(5):719-723.
[8]LuoY,Yin W,Signore AP,et al.Neuroprotection against focal ischemic brain injury by the PPARγagonist rosiglitazone[J].Neurochem,2006,97(2):435-448.
[9]Padilla J,Kaur K,Cao HJ,et al.Peroxisomel proliferator activator receptor-gamma agonists and 15-deoxy-Delta12,14-PGJ2 induce apoptosis in normal and malignant B-lineage cells[J].JImmuno,2000,16(5):6941-6948.
[10]Harris SG,Phipps RP.The nuclear receptor PPARgamma is expressed by mouse Tlymphocyte and PPARgamma agonists induce apotosis[J].EurJImmunol,2001,31(2):1098-1105.
[11]Shimazu T,Inoue I,Araki N,et al.PPARγagonist reduces infarct size in transient but not in permanent ischemia[J].Stroke,2005,36(2):353-359.
