氟比洛芬有关物质的合成
2013-03-08杨红光王庆河王亚丽马文希程卯生
杨红光,王庆河,王亚丽,马文希,程卯生
(沈阳药科大学 基于靶点的药物设计与研究教育部重点实验室,辽宁 沈阳110016)
(Key Laboratory of Structure-Based Drug Design &Discovery(Shengyang Pharmaceutical University),Ministry of Education,Shengyang 110016,China)
氟比洛芬(flurbiprofen)化学名为2-(2-氟-4-联苯基)丙酸,是由英国布茨公司开发的非甾体抗炎镇痛药。1976 年在英国上市,已列入英国、美国、欧洲、日本药典,临床上用于类风湿性关节炎、变节性关节炎、强制性脊椎炎等的治疗。氟比洛芬的消炎镇痛作用强,半衰期较长,服用剂量在苯丙酸类药物中最小。在氟比洛芬的合成中会有一些副产物生成,对其进行合成研究,有利于提高氟比洛芬的用药安全性,为此本文作者对氟比洛芬的有关物质进行了合成。
1 合成路线
有关氟比洛芬的合成方法,文献报道较多。近些年对其合成工艺改进的报道[1-2]多以2-氟-4-溴联苯为起始原料,经格氏反应制得格氏试剂,再与2-溴丙酸乙酯偶联得到中间体2-(2-氟-4-联苯基)丙酸乙酯,随后碱水解得到氟比洛芬。据欧洲药典记载,氟比洛芬的质量控制要求对(2RS)-2-(4-联苯基)丙酸(Ⅰ)、1-(2-氟-4-联苯基)乙酮(Ⅱ)、2-氟-4-联苯基羧甲酸(Ⅲ)进行含量测定,而目前国内尚无相应的对照品供应,因此对上述杂质进行合成研究具有重要意义。
Grigg 等[3]对(2RS)-2-(4-联苯基)丙酸(Ⅰ)的合成方法做了报道:以4-溴联苯为起始原料,经Suzuki 反应,Cu 催化下的硼氢化、羧化反应,最终得到Ⅰ。此合成反应过程中使用了过渡金属催化剂,价格昂贵,且硼化物毒性大。本文作者参考氟比洛芬的合成方法,经过优化,以4-溴联苯为起始物,经Grignard 反应制得相应格氏试剂,再与2-溴丙酸乙酯交叉偶联、碱水解得到目标化合物。此方法具有后处理简单、原料易得、成本低等特点。Eknath 等[4]对1-(2-氟-4-联苯基)乙酮(Ⅱ)的合成方法进行了报道,其中采用高价碘氧化试剂,反应时间长,成本较高。2-氟-4-联苯基甲酸(Ⅲ)的合成[5-6]与氟比洛芬的合成方法一致。鉴于目标化合物Ⅱ和Ⅲ有相同的母核,本文作者均以2-氟-4-溴联苯为起始原料,经Grigand 反应,分别与乙酸酐和二氧化碳反应制得相应的目标产物。氟比洛芬有关物质(Ⅰ、Ⅱ、Ⅲ)的合成路线见图1。
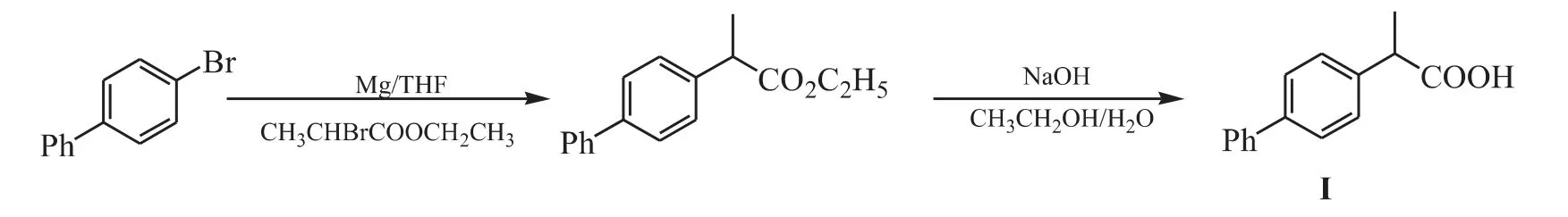
Figure 1 The synthetic routes to the target compounds

Continued Figure 1
2 合成实验
熔点采用Büchi B -540 熔点测定仪测定(温度未经校正);核磁共振氢谱采用Bruker ARX -300 型核磁共振仪测定(TMS 为内标);质谱采用Agilent 1100 LC -MSD -Trap -SL 质谱仪测定。薄层色谱法用硅胶板由青岛海洋化工厂生产;反应所用试剂均为国产分析纯或化学纯试剂。
2.1 (2RS)-2-(4-联苯基)丙酸(Ⅰ)的合成
向干燥的反应瓶中加入0.53 g(23.1 mmol)镁屑、80 mL 四氢呋喃(4 Å 分子筛干燥)、一粒碘,将溶于30 mL 四氢呋喃中的4-溴联苯5 g(22 mmol)加入滴液漏斗中,整个反应体系用氩气保护。回流状态下滴加少量4-溴联苯的THF溶液(5 mL),回流反应至碘颜色消失,继续滴加剩余原料4-溴联苯。待镁屑反应完毕,降至室温,滴加2-溴丙酸乙酯3.03 mL(22 mmol),回流反应2 h。蒸除THF,加入20 mL 0.52 mol·L-1稀硫酸淬灭反应,搅拌30 min,石油醚萃取(30 mL ×3),收集有机相,减压蒸除溶剂,得到的油状物溶于30 mL 乙醇中,滴加氢氧化钾溶液(2.22 g 氢氧化钾,15 mL 水),滴毕,回流反应1 h。冷至室温,蒸除乙醇,水层用浓盐酸调pH 值为2 ~3,有固体析出,抽滤,50 mL 石油醚(bp 60 ~90 ℃)重结晶,得白色固体(Ⅰ)3.22 g,总收率64.8 %,mp 137.9 ~140.1 ℃。1H-NMR(300 MHz,DMSO-d6)δ:12.35(1H,s,COOH),7.65(2H,d,J=1.3 Hz,Ph-H),7.62(1H,s,Ph-H),7.59(1H,s,Ph-H),7.46(2H,t,J=7.2 Hz,Ph-H),7.37(3H,m,Ph-H),3.72(1H,q,CH),1.39(3H,d,J=7.1 Hz,CH3)。ESI-MSm/z:249.1[M +Na]+。
2.2 1-(2-氟-4-联苯基)乙酮(Ⅱ)的合成
向反应瓶中加入0.5 g(21 mmol)镁屑、70 mL 四氢呋喃(4 Å 分子筛干燥)、一粒碘,将溶于50 mL THF 的2-氟-4-溴联苯5 g(20 mmol)加入滴液漏斗中,反应体系用氩气保护。回流下滴加少量2-氟-4-溴联苯的THF 溶液(10mL),待碘的颜色消失时滴加剩余原料。镁屑反应完毕后降至室温,转移至冷肼,于-78 ℃缓慢滴加乙酸酐1.89 mL(20 mmol),滴毕室温反应3 h。减压蒸除THF,加入30 mL 0.52 mol·L-1稀硫酸淬灭反应,乙酸乙酯萃取(30 mL × 3),无水硫酸钠干燥过夜。减压蒸除乙酸乙酯,得黄色蜡状产物,柱色谱分离得3.16 g 白色固体,收率73.8 %,mp 96.1 ~98.0 ℃。1H-NMR(300 MHz,DMSO-d6)δ:7.85(2H,m,Ph-H),7.69(1H,t,J=8.0 Hz,Ph-H),7.60(2H,m,Ph-H),7.49(3H,m,Ph-H),2.62(3H,s,CH3)。ESI-MSm/z:215.0[M +H]+。
2.3 2-氟-4-联苯基羧酸(Ⅲ)的合成
向反应瓶中加入1 g(42 mmol)镁屑、120 mL四氢呋喃(4 Å 分子筛干燥)、一粒碘,将溶于50 mL THF 的2-氟4-溴联苯10 g(40 mmol)加入滴液漏斗中,反应体系用氩气保护。回流下滴加少量2-氟-4-溴联苯的THF 溶液(10 mL),待碘的颜色消失时滴加剩余原料。镁屑反应完毕后降至室温,转移至冷肼,-20 ℃下通入二氧化碳气体20 min,室温搅拌4 h。减压蒸除THF,加入40 mL 0.52 mol·L-1稀硫酸淬灭反应,乙酸乙酯萃取(30 mL × 3),无水硫酸钠干燥过夜。减压蒸除乙酸乙酯,得类白色固体,二氯甲烷重结晶得7.52 g 白色针状结晶,收率87.0 %,mp 223.4 ~225.9 ℃。1H-NMR (300 MHz,DMSO-d6)δ:13.33(1H,s,COOH),7.85 (1H,dd,J= 8.0,1.4 Hz,Ph-H),7.75(1H,dd,J=8.0,1.4 Hz,Ph-H),7.65(1H,t,J=8.0 Hz,Ph-H),7.59(2H,m,Ph-H),7.49(3H,m,Ph-H)。ESI-MSm/z:214.8[M -H]-。
[1] 王尊元,马臻,梁美好,等. 氟比洛芬的新法合成[J].齐鲁药事,2005,24(11):687 -688.
[2] 蔡汉兴,朱明生,邱春明,等.2-(3-氟-4-苯基)苯丙酸合成[J].江西化工,2006,3:83 -86.
[3] GRIGG R D,RIGOLI J W,HOVELN R V,et al.Beyond benzyl grignards:facile generation of benzyl carbanions from styrenes[J]. Chem Eur J,2012,18(30):9391 -9396.
[4] BELLALE E V,BHALERAO D S,AKAMANCHI K G.Oxidative conversion of α,α-disubstituted acetamides to corresponding one-carbon-shorter ketones using hypervalent iodine reagents in combination with tetraethylammonium bromide[J].J Org Chem,2008,73(23):9473 -9475.
[5] NAGARATHNAM D,ASGARI D,SHAO J X,et al.Rho-kinase inhibitors:US,0208659[P].2001-03-21.
[6] CAPET M,LEVOIN N,BERREBI-BERTRAND I,et al.Novel dicarboxylic acid derivatives as S1P1 receptor agonists:EP,2008057571[P].2008 -06 -15.
