聚酯型聚氨酯多孔膜的制备和表征
2013-01-29王海军赵庭山刘战勋白林玉王学川
王海军, 赵庭山, 张 帅, 刘战勋, 白林玉, 王学川
(陕西科技大学 教育部轻化工助剂化学与技术重点实验室, 陕西 西安 710021)
0 引言
聚氨酯(Polyurethane, PU)是一种软硬段交替构成的嵌段共聚物,具有良好的渗透性,成为研究的热点膜材料[1, 2].聚酯型的PU氢键化程度较聚醚型的PU高,使得聚酯型的PU的机械性能优于聚醚型PU.此外,研究表明聚酯型PU膜具有比聚醚型更优的综合分离性能,优化聚酯型PU膜的制备工艺有望获得更大的应用价值.但是目前国内外的相关研究大多集中于合成新型的化学结构,如软段聚酯的结构组成、分子量以及原料摩尔比等因素对PU抗张强度等物性的影响;而制膜工艺和致孔剂对聚酯型PU膜微观结构的影响还缺乏系统性的研究[3-6].
本文考察了聚氨酯浆料的浓度、凝固浴的浓度和温度以及致孔剂等因素对聚氨酯薄膜形态结构的影响规律,并探讨了聚氨酯薄膜的微观结构与其性能的关系,研究结果对聚酯型聚氨酯分离膜的深入研究和应用具有借鉴意义.
1 实验部分
1.1 材料与设备
4,4-二苯基甲烷二异氰酸酯(MDI),分子量为250.26 g/moL,化学纯,烟台万华聚氨酯股份有限公司;聚己二酸丁二醇酯二醇(PBA),分子质量为3.5×104g/mol,化学纯,美国西格玛奥德里奇公司;1,4-丁二醇(BDO),分析纯,天津市福景化学试剂厂;聚乙二醇(PEG),分子质量为1 500 g/moL,化学纯,西安罗森伯科技有限公司;N,N-二甲基甲酰胺(DMF),分析纯,天津市风船化学试剂科技有限公司.
1.2 实验步骤
以硬段MDI为基础,分别加入不同的软段和扩链剂,3种工艺分别为加入软段PBA,扩链剂BDO,编号为1#;软段PBA,扩链剂PEG编号为2#;软段BDO,扩链剂PEG,编号为3#来合成聚氨酯.
合成工艺:将软段材料放入烘箱中加热熔融后,加入到带搅拌装置的洁净干燥的三口反应釜中,再加入一定量的DMF溶剂,并加热、搅拌,使其混合均匀.投入10.0 g的MDI,待其完全溶解后缓慢升温至75 ℃左右,预聚1.5 h.反应过程中连续搅拌,转速控制在400 r/min.预聚完成后,将扩链剂和部分DMF溶剂以及剩余的MDI加入到反应釜中,温度控制在80 ℃左右.根据反应物的黏度,补加适量的MDI调节体系的黏度在适当的范围.扩链反应进行1 h后,加入二月桂酸二丁基锡(DBTDL)催化剂,继续反应0.5 h,加入剩余的DMF,并用甲醇进行封端,20 min后终止反应得到最终产物[7].
多孔膜的制备:在产物中适当添加DMF控制聚氨酯浆料浓度,随后将聚氨酯混合浆料涂敷于洁净的玻璃板之上,使用0.5 mm的刮刀控制浆料厚度.再将玻璃板置入不同浓度和温度的凝固剂中,凝固完成后取出薄膜置于烘箱内干燥1.5 h,取出备用.
1.3 测试方法
测定聚氨酯的固含量及吸水率.采用Brookfieled公司DV-11+Pro数显型粘度计测定聚氨酯的黏度,转速设定为6 rpm.将制备的薄膜制成哑铃形的样品,使用INSTRON公司5565型拉伸试验机测定聚氨酯多孔膜的力学性能,拉伸速度为50 mm/min.使用HITACHI公司S4800型扫描电子显微镜观察聚氨酯薄膜的截面形态结构,加速电压为0.5 ~ 30 kV,放大倍率×(20~10 000).使用透水汽性测试皿测试膜的透水汽性.
2 结果与讨论
2.1 聚氨酯成膜性的比较
由于3种聚合工艺得到的聚氨酯分子链化学结构差异较大,其成膜性和薄膜性能表现出不同特点.1#聚氨酯(工艺: MDI+PBA+BDO)具有良好的成膜性,薄膜的弹性好柔软度高;2#聚氨酯(工艺: MDI+PBA+PEG)所得到的膜强度较好,但弹性和柔软度要比1#的差很多,薄膜僵硬卷曲,外观也不均匀,部分区域透明;3#(工艺: MDI+BDO+PEG)聚氨酯基本无法成膜.聚氨酯成膜性的优劣主要受分子链内软硬段和扩链剂结构的影响,通过比较上述3种聚合工艺的聚氨酯成膜性发现,以PBA为软段、BDO为扩链剂合成出聚氨酯具有良好的成膜性,制备的薄膜也具有优异的力学性能.因此后续制备聚氨酯多孔膜选用了1#聚氨酯浆液.1#聚氨酯浆液的固含量在35%左右,黏度为2 650 MPa·s.
2.2 成膜条件对聚氨酯多孔膜的结构与力学性能的影响
2.2.1 聚氨酯浆料的浓度
聚氨酯浆料的浓度是其凝固成型速度的主要决定因素,对多孔膜的截面结构影响很大.如图1a所示,当聚氨酯浆液浓度为70%时,薄膜内部形成连续性的多孔结构;聚氨酯浆液浓度降低到60%时,海绵状的微孔增多,孔径变大(图1b);如图1c所示,浓度降低到50%时连续性的孔洞数量明显减少,孔密度降低,均匀性变差.PU浆料初始浓度的高低一般对溶剂的扩散速率影响不大,但会影响溶剂质量分数分布的均匀性,浆液浓度越高越有利于提高薄膜结构的均匀性.
从表1中可以看出,PU薄膜的力学性能随着浆料浓度的降低而变差.PU浆料浓度由70%降低到50%时,膜的拉伸强度由1.82 MPa下降到1.56 MPa,断裂伸长率由141.63%下降到129.92%.此外,多孔膜内连续性孔洞的数量和均匀性也会影响薄膜的透水汽性.如表1所示,当PU浓度由70%降低至60%,膜的透水汽性由101增加到122 mg/(10 cm2×24 h);但当PU浆料浓度降低至50%时,膜的透水汽性明显降低.上述薄膜性能的差异是由其微观结构决定的.当PU浆液浓度较大时,膜内多为连续性的孔且孔密度高,薄膜的透水汽性好;当PU浆液浓度较低时,形成的多为封闭型的孔洞,导致其透水气性较差.要获得强度、断裂伸长率和卫生性能等综合性能优异的聚氨酯多孔膜,聚氨酯浆料浓度控制在60%以上.

图1 不同聚氨酯浆液浓度制备的薄膜截面的SEM照(PU浆料浓度: a=70%; b=60%; c=50%)
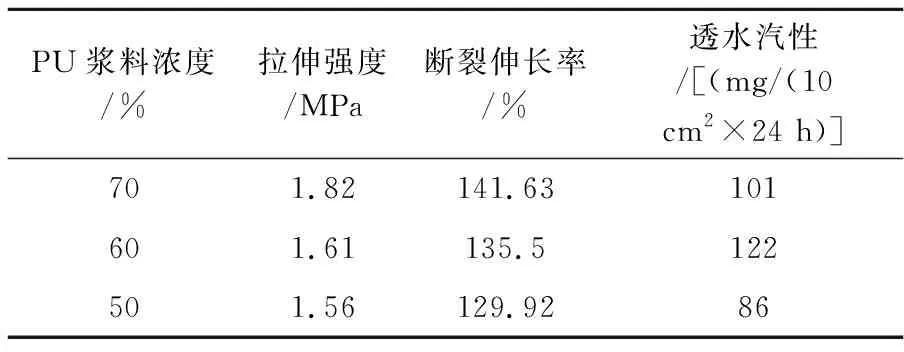
表1 不同PU浆料浓度成膜的物理性能
2.2.2 凝固浴的温度和浓度
凝固浴的温度和浓度是湿法成型过程中重要的工艺参数,直接决定着溶剂的双扩散速度和凝固过程速度.因此,通过调节凝固浴的温度和浓度,可控制凝固过程中溶剂的扩散速度,有利于调控薄膜的微观结构和宏观性能.图2为25 ℃、30 ℃和35 ℃的凝固浴中聚氨酯薄膜的微观结构扫描电镜图.如图2a所示,凝固浴温度较低时(25 ℃),薄膜内部结构均匀,多为指形微孔,且孔径较大.将凝固浴的温度提高到30 ℃后,薄膜表面形成了致密层.这是由于凝固浴温度的增加,涂覆在玻璃板上的聚氨酯浆料中的溶剂向凝固浴扩散的速度加快,致使凝胶分相迅速而形成致密层.当凝固浴温度升至35 ℃时,更大的溶剂扩散速率导致薄膜内部形成尺寸约为50μm的大孔.聚氨酯薄膜内部微观结构的变化导致其力学性能也发生相应的改变.如表2所示,随着凝固浴温度的升高,薄膜的力学性能明显提高,拉伸强度由2.16 MPa增加到2.53 MPa,断裂伸长率由141%增加到158.7%;但透水汽性则变化不大.

图2 不同凝固浴温度下聚氨酯成膜的截面SEM照片(a:25 ℃;b:30 ℃;c:35 ℃)

表2 不同凝固浴温度下聚氨酯成膜的物理性能
图3为不同凝固浴浓度下获得的聚氨酯薄膜的微观结构扫描电镜图.可以看出,随着凝固浴浓度的提高,薄膜内孔洞尺寸的均匀性变差.如图3a所示,当凝固浴浓度较低时(25%),形成长度约为5μm的指形孔,同时也分布有少量长度约为15μm的大孔,两种孔洞的孔径分布较为均匀,说明较低的凝固浴浓度有利于形成结构均一的多孔膜.如图3b和3c所示,随着凝固浴浓度的提高,孔洞数量明显减少,薄膜的致密度增大.

图3 不同浓度凝固浴中聚氨酯成膜的截面SEM照片(DMF/H2O: a=15%; b=20%; c=25%)
2.2.3 致孔剂
如图4所示,致孔剂的种类对聚氨酯薄膜的内部结构影响很大.体系中加入PEG后聚氨酯膜的孔径变小,孔密度较大(图4a,4c);但体系中加入BDO后薄膜的多孔结构更显著,孔径变大,但孔密度减小(图4b,4d).可以看出,体系中加入PEG或者BDO均可以起到了致孔剂的作用,在成膜过程中溶解于水而形成多孔结构.但由于BDO相对较低的分子质量使得成膜多孔结构的孔径较大,孔密度较小.
表3聚氨酯膜的力学性能测试数据表明,当PEG的加入量由5%增加到20%的过程中,聚氨酯膜的拉伸强度、断裂伸长率和透水汽性均有所增大;但过量的PEG会导致聚氨酯薄膜的力学性能下降.这是加入适量的PEG使得孔密度增加,膜的结构紧密,膜的力学性能提高;但过量的PEG造成膜的孔密度过大从而导致其机械性能下降.添加BDO对聚氨酯薄膜的影响与PEG类似,结果表明添加10%的BDO能够改善其力学性能,但过量后由于孔结构过大而导致薄膜力学性能变差.

图4 添加20%致孔剂后聚氨酯薄膜截面的SEM照片(a, c)为PEG;(b, d)为BDO
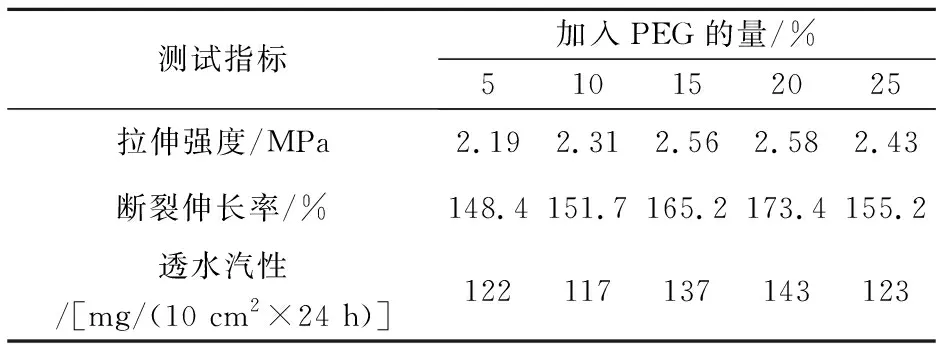
表3 加入致孔剂的聚氨酯成膜的物理性能
3 结论
用MDI做为硬段物质、PBA做为软段物质、BDO做为扩链剂合成的溶剂型聚氨酯具有良好的成膜性能.聚氨酯浆料的浓度、凝固浴的浓度和温度以及致孔剂等因素决定聚氨酯薄膜的结构与力学性能.聚氨酯浆料浓度控制在60%以上、凝固浴温度为30 ℃、凝固浴浓度要低于10%、添加适量的PEG或BDO等致孔剂,可获得结构均匀、孔径分布合理且力学性能优异的聚酯型聚氨酯多孔膜,具有良好的应用前景.
[1] Hasan E A,Cosgrove T,Round A N.Nanoscale thin film ordering produced by channel formation in the inclusion complex ofα-cyclodextrin with a polyurethane composed of polyethylene oxide and hexamethylene[J].Macromolecules,2008,41(4):1 393-1 400.
[2] Blit, Patrick H, Yi-Hao Shen, et al. Bioactivation of porous polyurethane scaffolds using fluorinated RGD surface modifiers[J].Journal of Biomedical Materials Research-Part A, 2010, 94(4):1 226-1 235.
[3] Zhou L J,Yu L Q,Ding M M,et al.Synthesis and characterization of PH-sensitive biodegradable polyurethane for potential drug delivery applications[J].Macromolecules,2011,44(4):857-864.
[4] Mohammadi T,Moghadam M T,Saeidi M,et al.Acid gas permeation behavior through poly(ester urethane urea) membrane[J].Ind.Eng.Chem.Res.,2008,47(19):7 361-7 367.
[5] Torini L,Argillier J F,Zydowicz N.Interfacial polycondensation encapsulation in miniemulsion[J].Macromolecules,2005,38(8):3 225-3 236.
[6] Castagna A M,Fragiadakis D,Lee H,et al.The role of hard segment content on the molecular dynamics of poly(tetramethylene oxide)-based polyurethane copolymers[J].Macromolecules,2011,44(19):7 831-7 836.
[7] Džunuzoviс' J V,Pergal M V,Poręba R,et al.Studies of the thermal and mechanical properties of poly(urethane-siloxane)s cross-linked by hyperbranched polyesters[J].Ind.Eng.Chem.Rev.,2012,51(33):10 824-10 832.
