端羟基P(GA-b-AMCMO)共聚醚的合成与性能①
2013-01-16卢先明刘亚静莫洪昌姚逸伦李大鹏
卢先明,刘亚静,莫洪昌,栗 磊,姚逸伦,李大鹏
(西安近代化学研究所,西安 710065)
0 引言
叠氮粘合剂具有高能、燃烧快、燃气清洁、热稳定性好等特点,是高能低特征信号推进剂和燃气发生剂的理想粘合剂[1-5]。叠氮粘合剂的典型代表为叠氮缩水甘油醚(GAP),但GAP基推进剂一直存在力学性能较差的问题,除了GAP均聚物存在分子侧链—CH2N3较多,链承载系数较小,数均相对分子质量Mn偏低等因素之外[2,6],一个重要原因是在研推进剂所用的含能增塑剂多为硝酸酯增塑剂,其与叠氮类聚合物在结构上存在较大差异、二者混溶性较差[7]。
为了改善叠氮粘合剂与硝酸酯增塑剂的混溶性,Manser等在叠氮粘合剂分子结构中引入硝酸酯基进行修饰改性,设想依据相似相溶的原理来改善其与硝酸酯增塑剂的混溶性。然而,硝酸酯基团的引入对力学性能的改善并不明显[7-9]。Hinshow在惰性粘合剂的分子链中引入—CN基。研究发现,此聚合物与硝酸酯增塑剂有着良好的混溶性[10],但遗憾的是该粘合剂不含叠氮基团,使推进剂的比冲损失过大。
本研究以1,3,5-三羟乙基异氰脲酸酯(THEIC)取代1,4-丁二醇为起始剂,经环氧氯丙烷(ECH)开环聚合和叠氮化取代等反应,首先制成带有氮杂环结构的Mn为2 500~3 000的三官能度叠氮缩水甘油醚(TGAP)[11-12],然后以其为大分子起始剂,引发含—CN基叠氮单体3-叠氮甲基-3-氰乙氧基甲基氧丁环(AMCMO)进行聚合,制备出一种新型嵌段叠氮粘合剂P(GA-b-AMCMO)[13]。通过在叠氮粘合剂分子结构中引入强极性的氮杂环结构和氰基功能基团[12-13],大幅提高了其与硝酸酯增塑剂混溶性能和相应叠氮推进剂的力学强度。
1 实验
1.1 原材料和仪器
原材料:ECH、N,N-二甲基甲酰胺(DMF)、1,2-二氯乙烷、二氯甲烷,分析纯,天津市科密欧化学试剂开发中心生产;GAP(Mn=2 850,羟值 =36.42 mgKOH/g)、AMCMO[13]为实验室自制;异氟尔酮二异氰酸酯(IPDI),德国拜尔公司生产;三氟化硼乙醚(BF3·OEt2),浙江省黄岩合成化工厂,用前重蒸;THEIC,常州市华安精细化工厂;无水NaCO3、异丙醇和石油醚(60~90℃)均为分析纯,西安化学试剂厂。
仪器和测试条件:用美国Nicolet公司60SXR-FTIR仪(KBr)和德国Bruker公司AVANCE AV500型核磁共振仪(1H-NMR,13C-NMR)对GA-b-AMMO共聚醚等聚合物的分子结构进行表征;用美国TA公司DSC910S差示扫描量热仪测量聚合物的Tg和热分解温度;羟值用上海精密科学仪器有限公司ZDJ-4A型自动电位滴定仪测定;聚合物的Mn采用英国PL公司GPC-50型凝胶渗透色谱仪测试,色谱柱为PLgel M IXED-E串联色谱柱,以PEG为标样,流动相为THF,柱温40℃;聚合物的粘度采用美国Brookfield公司CAP2000+椎板粘度计测定,测试温度50℃;推进剂的力学性能采用美国Instron公司Instron 6022型万能材料试验机测试。
1.2 分析测试方法
羟值采用吡啶-醋酐法,按照 Q/AY001—2002标准测定。力学性能用Instron 6022型万能材料试验机测试,采用GB/T 528—1992标准。
1.3 实验过程
1.3.1 合成路线
P(GA-b-AMCMO)的合成路线设计见下反应式,其中 X+Y+Z=20~30,X'+Y'+Z'=10~20,均为整数。

1.3.2 氯化聚醚多元醇CTP的合成
在装有搅拌、回流冷凝管、温度计和滴液漏斗的100 ml四口瓶中,加入起始剂 THEIC 4.698 g(0.018 mol)、二氯乙烷40 ml。搅拌升温至40℃,加入0.365 ml(0.002 9 mol)BF3·OEt2,待分散均匀后,滴加49.95 g(0.54 mol)ECH,以ECH滴加速度控制反应温度在30~40℃之间,滴完后继续室温反应24 h。反应完毕后,先用质量分数为0.7%的Na2CO3水溶液30 ml中和洗涤,分出有机相用质量分数为5%的盐水洗涤2次,每次30 ml;然后用蒸馏水洗涤1~3次直至中性,分出有机相减压蒸除溶剂得淡黄色粘稠透明液体CTP 53.12 g,收率为 97.2%。IR(KBr,ν,cm-1):3 449(—OH),1 693,1 463,764(THEIC 的氮杂环结构),1 126(C—O—C),749,706(C—Cl);1H—NMR(500 MHz,CDCl3,δ):3.6(CH2Cl),3.7(OCH2CH(CH2Cl)O);GPC数均分子量 MnGPC=2 695;羟值 =59.95 mgKOH/g;平均官能度 f=2.88;氯含量 34.47%;Tg=-38.23 ℃;50 ℃时粘度为19.14 Pa·s。
1.3.3 大分子起始剂TGAP的合成
在装有搅拌、回流冷凝管、温度计的100 ml三口烧瓶中,加入DMF60 ml和氯化聚醚多元醇CTP 31.8 g(0.011 8 mol),搅拌升温到60℃,分批加入叠氮化钠24.5 g(0.378 mol),加毕后升温到90 ℃反应48 h。反应完成后,趁热过滤除去固体杂质,减压蒸出大部分的DMF,加入50 ml二氯甲烷溶剂。然后,每次用50ml水洗除残存的DMF,共洗涤7~8次。分出有机相减压蒸除溶剂得淡黄色到淡红色粘稠透明液体TGAP 32.32 g,收率为 95.3%。IR(KBr,ν,cm-1):3 474(—OH),2 099,1 282(—N3),1 694,1 460,764(THEIC 的氮杂环结 构),1 126(C—O—C);1H-NMR(500 MHz,CDCl3,δ):3.4(CH2N3),3.7(OCH2CH(CH2N3)O);DSC热分解温度为256.38℃;MnGPC=2 784;羟值 =57.63 mgKOH/g;f=2.86;氮含量 38.69%;Tg=-43.90 ℃;50 ℃时粘度为0.79 Pa·s。
1.3.4P(GA-b-AMCMO)的合成
在装有搅拌、回流冷凝管、温度计和滴液漏斗的100 ml四口瓶中,加入大分子起始剂 TGAP 8.35 g(0.003 mol)和分散剂二氯甲烷15 ml。冷却到0℃后加入 BF3·OEt20.57 ml(0.004 5 mol),0.5 h 后滴加AMCMO 单体8.23 g(0.042 mol),约3~5 h滴加完毕后恒温反应70 h。反应完毕后先用质量分数为2%的Na2CO3水溶液15 ml中和洗涤,分出有机相后用质量分数为5%的盐水洗涤2次,每次15 ml;然后,用蒸馏水洗涤1~3次直至中性,分出有机相减压蒸除溶剂得黄色粘稠液体15.78 g,即 P(GA-b-AMCMO)粗品,收率为95.2%。
粗产物用石油醚与异丙醇组成的混合溶剂(体积比为1:1)溶剂萃取[11,14]。萃取剂与粗产物体积比为1:1,搅拌回流0.5 h,静止分层,待温度降到室温(25℃)时分出上层萃取液。再重复2次,脱去萃取剂得黄色粘稠透明液体14.03 g,总收率为84.6%。IR(KBr,ν,cm-1):3 483(—OH),2 251(—CN),2 102,1 281(—N3),1 693,1 461,764(THEIC 的氮杂环结构),1 111(C—O—C);MnGPC=5 089;羟值 =30.98 mgKOH/g;f=2.81;氮含量 33.56%;Tg= -38.51 ℃;DSC热分解温度为254.85℃;50℃时粘度为2.88 Pa·s。
2 结果与讨论
2.1 催化剂用量对AMCMO聚合反应的影响
催化剂用量过少,聚合反应难以引发或聚合不完全;催化剂用量过多则会导致聚合反应失控[11]。以MnGPC=2 784,f=2.86的 TGAP为大分子起始剂,BF3·OEt2为催化剂,通过核磁氢谱确定未聚合的游离单体与起始剂的摩尔比[15],在固定 nAMCMO/nTGAP=16/1,聚合温度0~3℃,3~5 h滴完单体后持续反应70 h的试验条件下,研究了催化剂用量对聚合反应的影响,其中,Mn理论值=5 920,f理论值=2.86。实验结果见表 1。
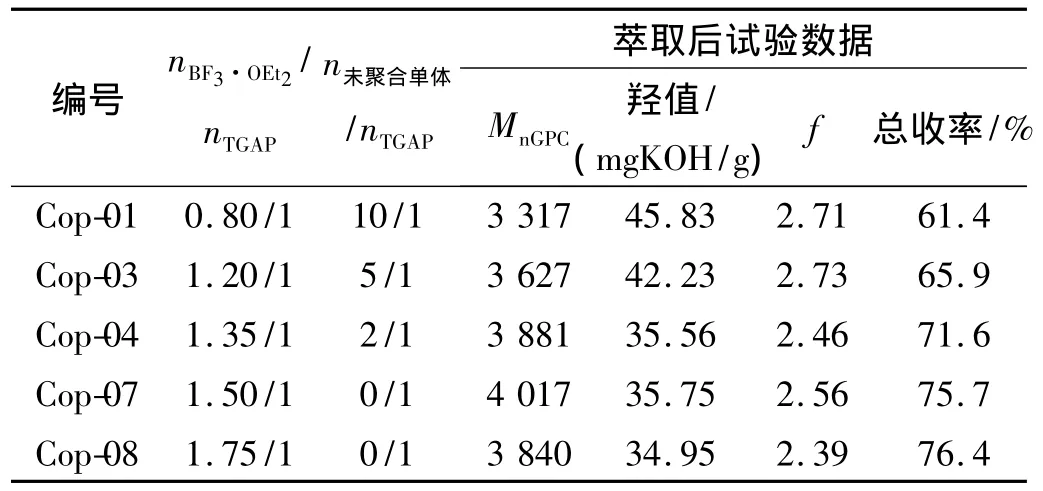
表1 催化剂用量对AMCMO聚合反应的影响Table 1 Effect of the proportion of catalyst on the polymerization
由表1可看出:(1)随着催化剂用量的增大,游离的单体愈来愈少,聚合进行得愈来愈完全,萃取后产品的总收率也愈来愈高。(2)当催化剂用量增大到nBF3·OEt2/nTGAP=1.50/1后,游离单体基本消失。此后,再增加催化剂用量,聚合的可控性开始变差。因此,确定催化剂用量为 nBF3·OEt2/nTGAP=1.50/1。
2.2 单体投料比对AMCMO聚合反应的影响
开环聚合理论表明阳离子聚合存在链转移,回咬成环等现象[11,16];文献[11]研究表明,环单体与小分子起始剂的比例越高,越易发生回咬和链转移;为探索大分子起始剂是否也遵循这一规律,在固定nBF3·OEt2/nTGAP=1.50/1,其他条件同上的试验条件下,研究了单体投料比对聚合反应的影响。其中,f理论值=2.86,结果见表2。
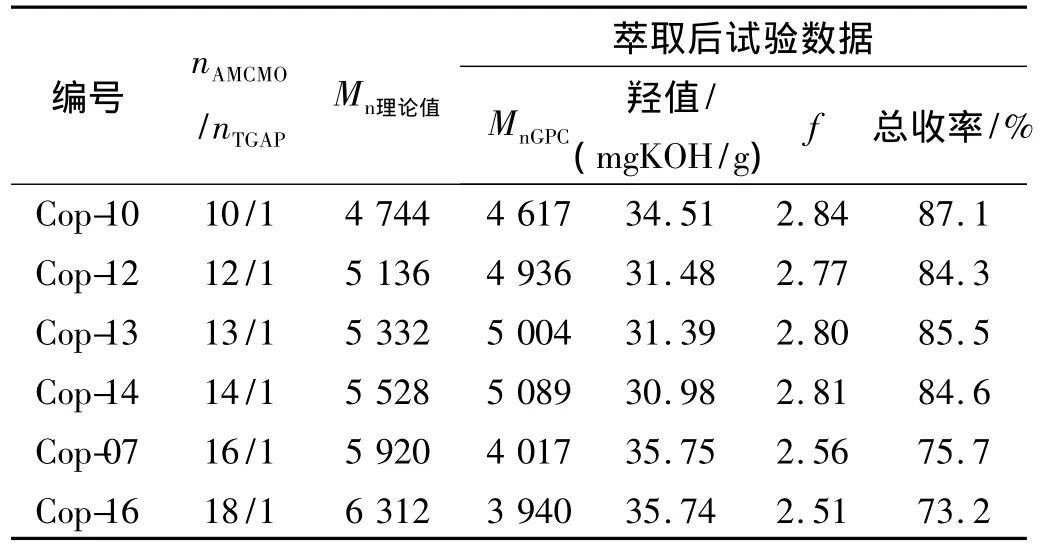
表2 单体投料比对AMCMO聚合反应的影响Table 2 Effect of the proportion of AMCMO on the polymerization
由表2可知:(1)当nAMCMO/nTGAP≥16/1时,Mn和f萃取后实测结果与理论值偏差较大,产品的总收率也较低;当nAMCMO/nTGAP≤14/1时,聚合反应较为可控,Mn和f萃取后实测结果则与相应理论值较为接近,产品的总收率也较高;这表明由于链转移,回咬成环等因素的影响,单纯增大单体比例并不会相应地提高P(GA-b-AMCMO)的Mn,即大分子起始剂引发环单体聚合同样存在可控聚合临界点的问题[11]。(2)当nAMCMO/nTGAP为13/1~14/1时,萃取后可获得 Mn≥5 000,f≥2.80的高分子量叠氮粘合剂P(GA-b-AMCMO),由此确定优化投料比为nBF3·OEt2/nAMCMO/nTGAP=1.5/13 ~14/1。
2.3P(GA-b-AMCMO)主链结构表征
P(GA-b-AMCMO)的主链结构可用1H NMR和13C NMR图谱来推测。图1和图2分别为均聚醚TGAP(nTHEIC/nGA实测值为1/26)与P(GA-b-AMCMO)(Cop-14,nTHEIC/nGA/nAMCMO理论值为 1/26/14)的1H NMR图谱,图3为 P(GA-b-AMCMO)(Cop-14)的13C NMR图谱。

图1TGAP的1H NMR图谱Fig.1 1H NMR spectrum of TGAP

图2 P(GA-b-AMCMO)的1H NMR图谱Fig.2 1H NMR spectrum of P(GA-b-AMCMO)
由图1、图2可知,化学位移δ约3.40处为GA链节中与—N3相连的—CH2的共振峰(共2个H原子)。图2与图1相比多出了δ2.60峰,该峰为AMCMO链节中与氰基相连的亚甲基上H原子的共振峰(共2个H原子)。P(GA-b-AMCMO)中GA与AMCMO的链节比可由 SGA/SAMCMO=Sδ3.40/Sδ2.60确定[15]。由图 2 可知,SGA/SAMCMO≈200.20/99.93≈26/13,与 nGA/nAMCMO理论值26/14较接近,进一步表明该投料比下聚合反应较为可控。
P(GA-b-AMCMO)各特征碳化学位移峰归属如图3所示,所有特征碳化学位移峰与P(GA-b-AMCMO)的结构均相对应。
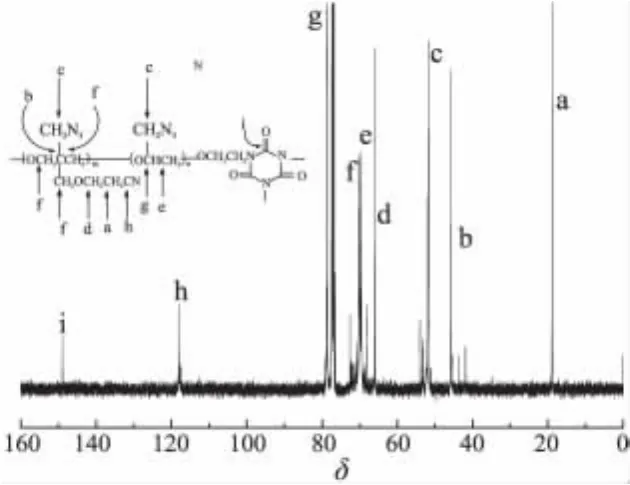
图3 P(GA-b-AMCMO)的13C NMR图谱Fig.3 13C NMR spectrum of P(GA-b-AMCMO)
2.4P(GA-b-AMCMO)与硝酸酯增塑剂的混溶性
将P(GA-b-AMCMO)与常用的硝酸酯增塑剂按比例配制成溶液,室温下静置,观察溶液在配制完毕4 d后的外观变化情况,其中NG/BTTN=1/1,结果见表3。

表3 P(GA-b-AMCMO)与硝酸酯增塑剂的混溶性Table 3 Compatibility of P(GA-b-AMCMO)with nitrate ester plasticizers
由表3可看出,P(GA-b-AMCMO)无论是在NG/BTTN还是在DEGDN中,当增塑比为2.5/1时,在4 d后P(GA-b-AMCMO)溶液仍然保持清亮不分层,这说明在叠氮粘合剂分子结构中引入强极性的氮杂环基团和氰基之后增加了聚合物的极性,提高了其与硝酸酯增塑剂的混溶性。
2.5P(GA-b-AMCMO)在Ls-2叠氮推进剂中应用
固定粘合剂/硝化甘油/铝粉/奥克托金/其他质量比为10/10/18/36/16/10,IPDI为固化剂的试验条件,将所合成的P(GA-b-AMCMO)(Cop-14)在Ls-2叠氮推进剂中进行系列应用研究,实验结果见表4。
由表4可见,与GAP相比,P(GA-b-AMCMO)基推进剂低温延伸率略有提高;高温拉伸强度则表现较好,最高可达0.86 MPa,约为GAP基推进剂的2~3倍。这表明通过在叠氮粘合剂分子结构中引入强极性的氮杂环结构和氰基功能基团,可大幅提高相应叠氮推进剂的力学强度。

表4 P(GA-b-AMCMO)推进剂的力学性能Table 4 Mechanical properties of propellants based on GAP and P(GA-b-AMCMO)(R=1.2)
3 结论
以Mn为2 500~3 000的三官能度端羟基叠氮均聚醚TGAP为大分子起始剂,AMCMO为单体,经阳离子聚合获得了一种Mn≥5 000,f≥2.80的新型高分子量叠氮粘合剂 P(GA-b-AMCMO)。与 GAP相比,P(GA-b-AMCMO)基推进剂表现出较好的力学强度,高温拉伸强度最高可达0.86 MPa,约为GAP基推进剂的2~3倍,表明通过在叠氮粘合剂分子结构中引入强极性的氮杂环结构和氰基功能基团,提高其与硝酸酯增塑剂的混溶性,可较大幅度地提高相应叠氮推进剂的力学强度。
[1] 龚士杰.GAP推进剂综述[J].推进技术,1991,14(1):67-70.
[2] 宋晓庆,周集义,王文浩,等.聚叠氮缩水甘油醚改性研究进展[J].含能材料,2007,15(4):425-430.
[3] Murali M Y,Padmanabha R M,Mohana R K.Synthesis,spectral and DSC analysis of glycidyl azide polymers containing different initiating diol unit[J].J.Appl.Polym.Sci.,2004,93(5):2157-2163.
[4] Murali M Y,Mani Y,Mohana R K.Synthesis of azido polymers as potential energetic propellant binders[J].Des.Monomers Polym.,2006,9(3):201-236.
[5] Kawamoto A M,Holanda J A S,Barbieri U,et al.Synthesis and characterization of glycidyl azide-r-(3,3-bis(azidomethyl)oxetane)copolymers[J].Propellant,Explos.Pyrotech.,2008,33(5):365-372.
[6] 曹一林,张九轩.四氢呋喃共聚型GAP粘合剂研究[J].固体火箭技术,1997,20(1):45-50.
[7] 甘孝贤,邱少君,卢先明,等.3-叠氮甲基-3-硝酸酯甲基氧丁环及其聚合物的合成及其性能[J].火炸药学报,2003,26(3):12-15.
[8] Manser G E,Malik A A,Archibald T G.3-azidomethyl-3-nitratomethyloxetane and polymers formed therefrom[P].WO05615,1994.
[9] Manser G E,Malik A A,Archibald T G.Polymers and copolymers from 3-azidomethyl-3-nitratomethyloxetane[P].USP 5463019,1995.
[10] Hinshow J C.Nitrate ester-miscible polyether polymers[P].USP 4804424,1989.
[11] 卢先明,甘孝贤,邢颖,等.端羟基聚环氧氯丙烷醚合成的可控性研究[J].聚氨酯工业,2009,24(5):15-18.
[12] Qiu Shao-jun,Gan Xiao-xian,Gao Chao,et al.Hydrogen bond effect of azido polyurethane elastomer by dynamic mechanical analysis[J].J.Polym.Sci.Part B:Polym.Phys.,2006,44(22):2841-2851.
[13] 甘孝贤,邢颖,李娜,等.3-叠氮甲基-3-氰乙氧基甲基氧丁环均聚物的合成与性能[J].火炸药学报,2004,27(4):10-13.
[14] 卢先明,甘孝贤,邢颖,等.端羟基聚环氧氯丙烷与叠氮聚醚的后处理研究[J].含能材料,2008,16(6):682-685.
[15] Tang Wei-yi,Huang Yan-gen,Qing Feng-ling.Synthesis and characterization of fluorinated polyacrylate graft copolymer capable as water and oil repellent finishing agents[J].J.Appl.Polym.Sci.,2011,119(1):84-94.
[16] 朱树新.开环聚合[M].北京:化学工业出版社,1987:41-42.
