分子筛限域孔道中吡啶的吸附结构和能量
2012-12-21褚月英郑安民
韩 冰 褚月英 郑安民 邓 风
(1中国科学院武汉物理与数学研究所,波谱与原子分子物理国家重点实验室,武汉核磁共振中心,武汉430071; 2中国科学院研究生院,北京100049)
分子筛限域孔道中吡啶的吸附结构和能量
韩 冰1,2褚月英1,2郑安民1,*邓 风1,*
(1中国科学院武汉物理与数学研究所,波谱与原子分子物理国家重点实验室,武汉核磁共振中心,武汉430071;2中国科学院研究生院,北京100049)
以吸附于ZSM-5孔道中的吡啶分子为例,利用量子化学理论方法考察了计算模型和密度泛函方法的选择对吡啶吸附结构和吸附能的影响,从而为准确计算分子筛限域孔道中客体分子吸附态结构和能量参数提供了依据.计算结果表明,吡啶吸附能随着所选用的分子筛的计算模型(从8T到128T)增大而增大,当选用的孔道结构能够将整个分子筛的孔道结构完全包括进来的时候(72T)达到收敛.与常规的密度泛函方法(B3LYP和M06-2X)相比较,考虑到色散作用校正的B97D泛函方法能够很好地处理分子筛体系中主客体间的长程相互作用和弱相互作用,计算得到的能量数据与实验结果符合得很好.
量子化学计算;分子筛;限域效应;吸附能
1 引言
分子筛催化剂因为环境友好而在石油化工中被广泛应用.作为固体酸催化剂,它具有不腐蚀设备、容易与液相反应体系分离、环境污染小、选择性高等特点.1由于分子筛具有独特的孔道特征、较好的热稳定性和优越的离子交换能力,2因此在裂解、质子化、异构化和聚合等反应中有广泛的应用.3分子筛的酸性特征直接与催化活性相关联,研究分子筛的表面酸性对于深入理解其催化活性及催化机理具有重要意义.4,5吡啶作为一种碱性探针分子被广泛地用来表征固体酸催化剂的酸强度,例如在程序升温吸脱附(TPD)方法中,通过比较吡啶脱附温度的高低来确定酸强度的大小;而在核磁共振(NMR)探针分子方法中,通过比较吡啶吸附前后的1H化学位移变化来定量测量酸强度.6通过吡啶的TPD和NMR测定方法,人们可以很直观地比较催化剂的酸强度大小,而这些谱学参数与吡啶在酸位表面的吸附构型和吸附热参数是密切相关的.7由此看来,研究吡啶与分子筛酸位的主客体相互作用的结构和能量(吸附热)参数对于在原子分子水平下解释实验谱学参数显得特别重要,然而这些重要信息并不能通过实验方法来直接获取.
理论计算的方法作为实验方法有力的补充,已经被广泛用来分析固体催化剂活性中心的结构和催化反应机理.彭少逸等8研究了B、Al、Ga等同晶取代进入丝光沸石骨架后可能存在的位置,考察了杂原子进入骨架对丝光沸石Brönsted酸性的影响,孙淮等9研究了磷在P-ZSM-5沸石中存在的形态, Yang等10研究了La离子在分子筛骨架中的存在形态和Lewis酸性特征,Zhou等11研究了钼掺杂的ZSM-5分子筛上甲烷脱氢芳构化反应路径.在众多的量子化学理论方法中,密度泛函理论(DFT)是目前应用最广泛的理论计算方法之一,该方法不需要很大的计算资源却得到不错的精确度,因此被广泛应用到有机体系热力学和动力学的预测.12,13
分子筛催化剂由于具有纳米量级的三维孔道结构而在石油化工领域有着广泛的应用,其独特产物的选择性源于客体分子(反应物、过渡态、产物或反应中间体)与分子筛限域孔道中的吸附或排斥作用.孔道限域效应主要表现为吸附分子与分子筛孔道之间的长程主客体相互作用(如范德华作用).众所周知,经典的DFT泛函并不能用来精确描述范德华作用力以及氢键这类弱相互作用,理论预测的吸附能往往较实验值偏低.14因此,建立新的理论方法来准确描述氢键和范德华作用力是当前理论研究的热点,其中Becke小组15在1998年开发出混合梯度近似泛函如B97系列,这种泛函对于研究短程作用为主的体系有较高的计算精度.后来,Truhlar等16于2006年研发了M06系列泛函(M06、M06-L、M06-2X),研究结果表明这一系列泛函能够更好地描述非键相互作用.这是因为色散力作为范德华作用力的主要来源,在研究分子间的相互作用时非常重要而不能被忽视.可以设想,如果在理论计算中能够将色散相互作用考虑进来,那么计算得到的吸附结构和吸附能参数将更接近于实验值.Grimme17于2004年提出了DFT-D算法,利用半经验的-C6/R6形式的色散项来表示范德华作用力.在DFT-D算法中,系统的总能量(EDFT-D)表示为DFT计算得到的能量(EDFT)和长程相互作用的色散能贡献(Edisp)之和,即:EDFT-D=EDFT+Edisp.其中长程相互作用的色散能贡献Edisp对于短程作用为零,则回归为DFT泛函的原始形式,不影响泛函本身在短程相关效应描述较好的原有优点.DFT-D解决了DFT泛函不能很好地描述弱相互作用及长程作用中的范德华作用的不足,因而引起了人们越来越多的兴趣.基于B3LYP而改进的B3LYP-D算法,是利用在B3LYP方法优化的基础上,在最后加上色散校正项而得到最终的单点能参数;同样B97-D方法与B3LYP-D方法类似,在单点能部分加上色散作用的校正.两种方法的优势还在于占用的计算资源不大,计算精度也比较高.而B97D方法则与B97-D方法有所不同,它是在结构优化的每一步都将色散相互作用考虑进来.18
准确描述有机物在分子筛孔道中的吸附行为的理论研究对于解释实验现象、揭示反应机理是非常重要的.目前B3LYP、M06-2X、B97D等方法广泛用于研究分子筛的酸性特征,预测分子筛催化机理和过渡态特征.19-22然而,哪种泛函方法得到的结构参数和吸附能与实验更为接近,文献一直没有相关的报导.所以有必要通过系统考察B3LYP、B971、M06-2X以及考虑色散校正作用的B3LYP-D、B971-D和B97D等6种理论方法的计算结果,与实验结果对比来确定能够准确预测有机物种在分子筛孔道中吸附结构和能量的泛函方法.本文拟从分子筛计算模型和理论方法的选择两个方面入手,探索出能够准确预测有机分子在分子筛孔道中结构参数和吸附能量的准确方法.
2 计算方法
2.1 模型的选取
为了考察分子筛孔道结构对Brönsted酸位和吡啶吸附的影响,分别选取8T、30T、46T、60T、72T和128T团簇模型来表示分子筛ZSM-5的孔道结构(见图1).需要指出的是,8T模型仅将Brönsted酸活性中心的局域结构考虑进来,随着模型的逐渐增大,分子筛完整的孔道结构将被重现出来.基于客体分子的易接近性以及酸位的稳定性,选取分子筛ZSM-5中Si12-O24(H)-Al12位点来表示其酸性中心.23在搭建模型中,末端Si原子用H原子来饱和,H原子的取向与原晶体中被取代的氧原子取向一致,并且所有末端Si-H键长设置为0.147 nm.
2.2 DFT计算方法
所有的理论计算都采用Gaussian 09软件包24完成.随着模型选择的增大,体系原子增多(例如128T模型,结构式为Si127Al1O219H75,共422个原子),对于如此庞大的体系如果选择单纯的DFT方法因计算量太大而在现有的计算水平下显然无法实现.为了提高计算效率,在这里采用分子力学和量子力学结合的ONIOM(our own N-layer integrated MO and MM method)方法,25,26该方法已成功应用于研究分子筛的催化反应机理.27在计算中,将体系分为两层,其中活性中心采用比较精确的量子力学方法(比如B3LYP、M06-2X和B97),体系中其余的部分则用较低水平的半经验(MNDO)方法加以处理.这样可以保证较好的计算精度,同时也能减小计算量,提高计算效率.在30T-128T簇模型体系中,高层为8T区域(SiO)3Si-OH-Al(OSi)3与吸附的吡啶分子,优化分别采用B3LYP、B971、B97D和M062X方法,基组为6-31G(d,p);剩余的部分为低层,采用MNDO半经验方法.在结构优化过程中,只将分子筛的活性中心部位(O3Si-OH-AlO3)与吡啶分子进行全优化,剩下的原子都被固定在原来的晶格位置,这样可以保持分子筛模型在结构优化的过程中不失去其特有的晶体结构.对于小的8T ZSM-5结构优化采用同样的密度泛函方法以及基组进行计算,在结构优化过程中对O3Si-OH-AlO3酸中心和吡啶分子进行优化,而末端-SiH3基团都固定在原来的晶体位置.单点能计算分别选用B3LYP、B971、B97D和M06-2X方法,基组为6-31G(d,p).而B971-D和B3LYP-D的单点能则是在B971和B3LYP方法得到的单点能基础上加上相应的Edisp校正.

图1 B3LYP/6-31G(d,p)水平下8T-128T ZSM-5模型优化得到的Brönsted酸位结构Fig.1 Optimized structures of Brönsted acid sites with 8T-128T ZSM-5 models at B3LYP/6-31G(d,p)level (a)8T,(b)30T,(c)46T,(d)60T,(e)72T(f)128T;distances in nm
2.3 色散相互作用(Edisp)的计算

Grimme17和Sumpter28提出长程相互作用的色散能贡献可以用公式(1)表示:在这里Nat为体系中的原子数,S6为校正系数,通常不同的泛函对应不同的校正系数,比如PBE的校正系数为0.75,B3LYP为1.05,B97D为1.25;17C6为原子的色散系数,其大小与原子的电离电势和静态偶极化率相关;fdamp(Rij)是与原子间距离Rij相关的阻尼函数,其表达式可以用公式(2)表示:其中R0为原子的半径,i和j分别为体系中i和j两个原子的原子序号,Rij为i和j两原子间的距离.表1给出了H、C、O、N、Al和Si原子的色散系数C6和原子半径R0等有关参数.25基于公式(1)和公式(2),色散相互作用(Edisp)的计算由本课题组编写的C程序完成.

3 结果与讨论

表1 色散能计算中H、C、N、O、Al和Si原子的色散系数(C6)和原子半径(R0)25Table 1 Dispersion coefficient(C6)and radii(R0)for elements H,C,N,O,Al,and Si in the calculations of dispersion energy25
3.1 分子筛酸性特征的理论研究
分子筛的酸性特征将直接影响到吸附的有机物种的结构和催化反应活性.van Santen29和Blowers30等研究了固体酸酸强度对烷烃活化反应(如氢交换、裂解、脱氢等)活化能垒的影响,发现催化活性随着酸强度的增大而显著增强.在前期的研究工作中,我们6,31,32发现固体酸的酸强度也将影响到探针分子的吸附和谱学参数,固体酸的酸性越强,酸性质子向吡啶、三甲基膦氧等碱性探针分子转移的程度就越显著,同时探针分子的化学位移也表现出规律性的变化.为了讨论固体酸模型大小对酸性中心的结构和酸强度的影响,基于8T、30T、46T、60T、72T和128T模型的计算得到的Brönsted酸中心的结构参数和质子的Mulliken电荷(QH)列于表2中.可以发现不论采用哪种泛函方法,随着模型的增大结构参数呈现出相同的变化规律.以B3LYP方法为例,随着模型的增大,30T、46T模型的Si-O键长分别从8T模型的0.1662 nm减小为0.1658和0.1654 nm,而Al-O键长则在0.1833 nm的基础上分别减小了0.0006和0.0013 nm,说明模型的增大(从8T到46T)对酸位的局域骨架结构有一定的影响;而46T增大到128T模型的结构参数变化则不明显,说明对于46T以上模型,其结构参数已经达到收敛,模型的进一步增大对骨架结构的影响很小.同样,对于羟基O-H键长,8T模型为0.0969 nm,30T和46T分别为0.0971和0.0973 nm,46T以上模型O-H键长维持在0.0973 nm不变;Q(H)即H的电荷从0.388|e|逐渐增加到0.401|e|后保持不变,同样在46T呈现收敛的趋势.

表2 不同计算方法下8T-128T ZSM-5模型去质子化能(DPE)、H的电荷(QH)和主要结构参数Table 2 Deprotonation energy(DPE),Mulliken charge for H(QH),and main geometry parameters of 8T-128T ZSM-5 models calculated with different methods
分子筛的催化反应活性是与其酸性特征密切相关的,通过rO-H和Brönsted质子QH的变化可以定性地观察酸强度的变化趋势.从表2给出的rO-H和QH的数据来看,分子筛的酸强度随着选用模型的增加而缓慢增强.除此之外,去质子化能(DPE)是一种有效表征酸强度大小的定量方法.33DPE定义为酸位脱去一个质子所要吸收的能量(AH→H++A-),在数值上表现为酸位(AH)的能量与失去质子后形成共轭碱(A-)的能量差值:DPE=E(A-)-E(AH).较小的DPE代表较强的酸性,在酸催化反应中质子转移就较容易.从表2中发现8T、30T、46T、60T、72T和128T的DPE计算值(由B3LYP方法计算得到)分别是1257.5、1256.2、1235.7、1235.6、1235.6和1235.6 kJ· mol-1,呈先降低后收敛的趋势;不同的固体酸模型的DPE值与ZSM-5分子筛的实验值范围1217.8-1255.5 kJ·mol-134接近.如表2所示,这种变化趋势在使用其他方法计算的结果中也有类似的体现.这说明分子筛的酸强度主要取决于酸性中心的局域结构,而孔道结构的影响不大.
3.2 不同计算模型研究吡啶在分子筛孔道中的吸附

图2 B3LYP/6-31G(d,p)方法下8T-128T ZSM-5模型吸附吡啶后优化得到的结构Fig.2 B3LYP/6-31G(d,p)optimized structures of pyridine adsorption complexes on 8T-128T ZSM-5 models (a)8T,(b)30T,(c)46T,(d)60T,(e)72T,(f)128T;distance in nm
分子筛孔道和被吸附分子之间的主客体相互作用将直接影响到吸附分子的构型以及其物理化学性质.被吸附分子与分子筛孔道之间主要是静电吸引和范德华作用,当被吸附分子逐渐被孔道包围时,这种稳定作用也逐渐增强,即“孔道限域效应”.在研究了不同大小的分子筛模型对其固有酸强度的影响之后,我们将进一步研究孔道效应对吡啶结构的影响.图2给出了B3LYP计算方法下ZSM-5不同酸模型的吡啶吸附结构图.比较图1和图2中分子筛和吡啶的构型变化后可以发现,在吡啶吸附前后rO-H键长的变化是非常明显的.以72T模型为例,吡啶吸附前,O-H键长为0.0973 nm,吸附吡啶后, O-H键长增大为0.1559 nm,而N-H键长为0.1045 nm,这表明吡啶被质子化形成了吡啶离子.相应的,吡啶吸附前后Brönsted酸位上的O原子与Si和Al的距离也呈现一定的趋势:对于8T模型, O-Al和O-Si键长分别为0.1833和0.1662 nm;在吡啶探针分子作用下,O-Al和O-Si键长分别减小为0.1749和0.1594 nm;对于72T模型,吡啶吸附前后O-Al和O-Si键长分别从0.1820和0.1653 nm减小为0.1744和0.1589 nm.表3给出了在不同模型和方法下计算得到质子的Mulliken电荷(QH)和结构参数.研究结果表明不论采用哪种泛函方法,随着模型的增大,Eads、QH以及结构参数都呈现出相同的变化规律.以B3LYP方法为例,随着酸模型的增大,N-H键长逐渐从0.1075 nm减小到0.1045 nm,而后者与气态吡啶离子的N-H键长0.1017 nm是比较接近的.另外通过比较不同模型间的参数变化发现,8T和30T模型间的参数变化为ΔrN-H=0.0026 nm,ΔrO-H=0.0005 nm;30T和46T之间变化很小ΔrN-H=0.0003 nm,ΔrO-H=0.0002 nm,而60T到128T之间则基本没有变化.值得注意的是,在8T模型中, QH为0.420|e|,而在72T模型中QH减小为0.400|e|.这说明随着酸模型的增大,越来越多的正电荷被分散到吡啶分子的共轭环中,增大了吡啶离子的稳定性.这也进一步说明了孔道结构对吡啶吸附起到了稳定的作用.不同方法计算得到的吡啶吸附参数有一定的差别,说明不同的计算方法对吡啶在分子筛表面吸附的构型是有影响的;但从8T到128T模型参数的变化趋势上看,无论哪一种泛函方法,其整体变化趋势都是一致的.

表3 不同计算方法下8T-128T ZSM-5模型吸附吡啶后H的电荷和主要结构参数Table 3 Mulliken charge for H and main geometry parameters of pyridine adsorption complexes on 8T-128T ZSM-5 models calculated with different methods
表4给出了吡啶在不同的酸模型表面上的吸附能.吸附能计算公式如下:
ΔEads=(EZOH+Epyridine)-Epyridine-ZOH(3)其中Epyridine-ZOH为复合物的能量,EZOH和Epyridine分别为分子筛模型和吡啶分子的单点能.在这里需要提出的是,计算得到的吸附能是体系在绝对零度的相互作用能,其值与实验得到的吸附热接近.35,36表4给出的吡啶吸附能结果分析发现,随着模型的增大,吸附能呈增大趋势并最后达到收敛.对于B3LYP方法,8T模型的吸附能为45.4 kJ·mol-1,它包含了酸位的局域结构,但没考虑孔道;30T和46T模型吸附能分别为85.4和88.9 kJ·mol-1,与8T模型相比吸附能分别升高了40和43.5 kJ·mol-1,这是由于30T和46T模型既包含了酸位的局域结构,又包含了部分孔道;60T、72T和128T模型的吸附能分别为90.0、90.4和90.6 kJ·mol-1,模型之间能量只有0.6 kJ· mol-1的变化,说明孔道结构已经逐渐完整,对于吡啶的吸附影响变小,能量达到收敛.这说明对于复杂的ZSM-5分子筛体系,72T模型可以完全将孔道对吸附分子之间的相互作用都包括进来,可以用来反映ZSM-5分子筛真实的孔道结构.对于其他方法计算得到的吡啶吸附能(表4)也同样表现出随着计算模型的增大而增大,在72T模型时能量达到收敛.
3.3 不同泛函方法研究吡啶在分子筛孔道中的吸附
基于优化得到的吡啶分子吸附结构,我们在表4中给出了不同模型和计算方法下吡啶在ZSM-5分子筛表面的吸附能.在前面的研究中我们已经确定了72T模型可以用来反映分子筛ZSM-5的孔道结构,所以在这里以72T模型为例比较不同计算方法得到的吸附能.在B3LYP/6-31G(d,p)水平下计算得到的吸附能为90.4 kJ·mol-1,而实验值为200 kJ· mol-1.37众所周知对于质子化的吡啶离子与分子筛孔道之间的相互作用,除了电子相关效应外,静电吸引和范德华作用也起到稳定吡啶离子的作用. B3LYP方法仅仅能对电子相关效应作出较好的描述,而不能够很好地描述范德华作用力,因此计算得到的吸附能与实验值误差很大.B971/6-31G(d,p)水平下计算得到的吸附能为110.4 kJ·mol-1,与B3LYP方法相比计算精度稍有提高,这是因为B97方法在电子相关方面进行了一定的修正,对于研究短程作用为主的体系有较高的计算精度,但由于对范德华作用力的描述依旧不是很全面,所以计算得到的吸附能与实验值仍有很大的误差.M06-2X方法考虑了弱相互作用力,在计算范德华作用方面性能比B3LYP有所改善,计算得到的吸附能量为144.3 kJ·mol-1,与B3LYP相比计算精确明显提高.19但对于主客体间普遍存在的长程色散作用,M06-2X未能体现出色散作用对吸附能量的影响,对于分子间的色散作用描述不是很充分,计算结果与实验值仍有约30%的误差.B3LYP-D和B971-D分别是在B3LYP和B971优化后对色散作用进行校正,因此得到的能量比B3LYP和B971方法计算的结果进一步提高,分别为122.4和122.5 kJ·mol-1,但这两种校正方法的最大问题在于仅仅在单点能计算部分增加了一项色散校正,并没有将弱相互作用的影响引入结构优化过程中,虽然相对于纯的密度泛函计算得到的能量有所改进,但是与实验值相差仍然约80 kJ·mol-1.B97D方法是在B97的基础上,每一步结构优化的过程中都进行色散校正,因此可以预期该种方法得到的结构和能量精度将会有很大的提高.从表3给出的不同方法下吡啶吸附的结构参数可以发现,B97D方法计算得到的N-H和O-H键长分别为0.1047和0.1563 nm,比B971方法计算得到的N-H键长增加了0.0002 nm,O-H键长减小了0.0004 nm;基于B97D水平下得到的结构参数,我们进一步计算得到其吸附能为204.7 kJ·mol-1,与实验值200 kJ·mol-1吻合得较好,这也进一步验证了B97D方法在计算色散作用力方面有较大的优势.38图3给出了不同团簇模型和不同方法下吡啶的吸附能以及实验值,从图中可以更直观地看出B97D方法与实验值吻合得最好.

表4 不同计算方法下得到的8T-128T ZSM-5模型吡啶吸附能(Eads)Table 4 Pyridine adsorption energies(Eads)on 8T-128T ZSM-5 models calculated with different methods
3.4 分子筛体系理论计算模型和方法的选择
分子筛催化剂的优越性主要体现在其具有一定的酸性和独特的孔道特征,因此在选择计算模型的时候需要充分考虑这两个因素.通过系统地研究发现分子筛的酸强度主要取决于酸性中心的局域结构,孔道结构对分子筛的固有酸强度影响不大.从图2给出的吡啶吸附结构图和表4给出的吡啶吸附能结果可以发现,随着模型选择的增大,孔道结构逐渐完整,“孔道限域效应”越来越明显,吸附能呈增大趋势并在60T-128T达到收敛.另外需要注意的是,随着模型的增大,体系原子增多,计算量也相应增大.基于以上因素考虑,我们选用72T模型来进行分子筛体系的理论计算,因为72T模型既能表现出ZSM-5的酸性特征,又具备完整的孔道结构,可以充分地反映ZSM-5的催化性能;另外72T模型原子数量适中,计算量比128T小,计算精度比小模型高,因此选用72T模型来代表ZSM-5分子筛的孔道结构应用于一些理论计算可以得到很好的结果.
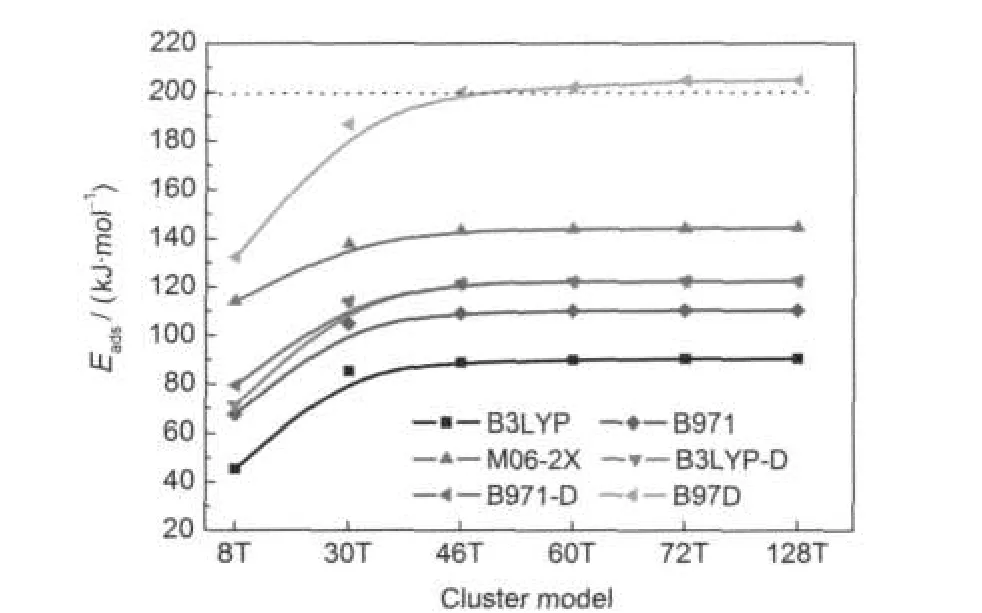
图3 六种计算方法下8T-128T ZSM-5模型吡啶吸附能Fig.3 Adsorption energies of pyridine on 8T-128T ZSM-5 models calculated with six different methods The experimental value(200 kJ·mol-1)37is shown by the dotted line.
分子筛体系中的主客体间相互作用非常复杂,除了电子相关效应外,静电吸引、范德华作用以及色散作用等长程相互作用对于有机物种在分子筛孔道中的吸附都有不同程度的影响.因此选择合适的计算方法用于准确描述有机物种在分子筛孔道中的吸附结构和能量,对于解释实验现象、揭示反应机理是非常重要的.通过研究不同泛函方法计算吡啶在分子筛孔道中吸附的结果发现,B97D方法可以很好地处理范德华作用等弱相互作用,在计算色散作用力方面也有较大的优势,是一种能够准确预测有机分子在分子筛孔道中结果参数和吸附能量的准确方法.
4 结论
通过量子化学计算的方法系统考察了计算中分子筛模型和泛函方法对分子筛固有酸强度与吡啶吸附结构和能量的影响.结果表明,分子筛的酸强度主要取决于酸性中心的局域结构.而在研究反应物在分子筛限域孔道中吸附和转化过程中,为了考察吸附分子和分子筛孔道之间的主客体相互作用,有必要采用大的模型将分子筛完整的孔道结构包括进来.由于经典的B3LYP泛函方法未能将色散作用力考虑进来,采用该方法计算的吸附能量明显低于实验值.为了获得精确的结构和能量数据, B97D泛函方法在结构优化的每一步都作出色散作用力的校正,计算结果与实验值能够很好的吻合.References
(1) Weitkamp,J.;Traa,Y.Catal.Today 1999,49,193.
(2) Slagtern,A.;Dahl,I.M.;Jens,K.J.;Myrstad,T.App.Catal.AGen.2010,375,213.
(3)Luzgin,M.V.;Parmon,V.N.Angew.Chem.Int.Edit.2008,47, 4559.
(4)Yu,Z.W.;Zheng,A.M.;Wang,Q.;Huang,S.J.;Deng,F.;Liu, S.B.Chin.J.Magn.Reson.2010,27,485.[喻志武,郑安民,王 强,黃信炅,邓 风,刘尚斌.波谱学杂志,2010,27,485.]
(5) Zheng,A.M.;Huang,S.J.;Deng,F.;Liu,S.B.Phys.Chem. Chem.Phys.2011,13,14889.
(6)Zheng,A.M.;Zhang,H.L.;Chen,L.;Yue,Y.;Ye,C.H.;Deng, F.J.Phys.Chem.B 2007,111,3085.
(7) Coma,A.Chem.Rev.1995,95,559.
(8)Yuan,S.P.;Wang,J.G.;Li,Y.W.;Peng,S.Y.Acta Physico-Chimica Sinica 2001,17,811.[袁淑萍,王建国,李永旺,彭少逸.物理化学学报,2001,17,811.]
(9)Yang,J.;Sun,Y.X.;Zhao,L.F.;Sun,H.Acta Physico-Chimica Sinica 2011,27,1823. [杨 静,孙迎新,赵立峰,孙 淮.物理化学学报,2011,27,1823.]
(10)Yang,G.;Wang,Y.;Zhou,D.H.;Zhuang,J.Q.;Liu,X.C.;Han, X.W.;Bao,X.H.J.Chem.Phys.2003,119,9765.
(11)Zhou,D.H.;Ma,D.;Liu,X.C.;Bao,X.H.J.Chem.Phys. 2001,114,9125.
(12) Hohenberg,P.;Kohn,W.Phys.Rev.1964,136,864.
(13)Kohn,W.;Becke,A.D.;Parr,R.G.J.Phys.Chem.1996,100, 12974.
(14) Kristyan,S.;Pulay,P.Chem.Phys.Lett.1994,229,175.
(15)Schmider,H.L.;Becke,A.D.J.Chem.Phys.1998,108,9624.
(16) Zhao,Y.;Schultz,N.E.;Truhlar,D.G.J.Chem.Theory Comput.2006,2,364.
(17)Grimme,S.J.Comput.Chem.2004,25,1463.
(18)Grimme,S.J.Comput.Chem.2006,27,1787.
(19) Zhao,Y.;Truhlar,D.G.J.Phys.Chem.C 2008,112,6860.
(20) Pidko,E.A.;Hensen,E.J.M.;van Santen,R.A.J.Phys.Chem. C 2008,112,19604.
(21) Boronat,M.;Martinez,C.;Corma,A.Phys.Chem.Chem.Phys. 2011,13,2603.
(22) Boekfa,B.;Choomwattana,S.;Khongpracha,P.;Limtrakul,J. Langmuir 2009,22,12990.
(23) Vankoningsveld,H.;Van Bekkum,H.;Jansen,J.C.Acta Crystallogr.B 1987,43,127.
(24) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, B.01;Gaussian Inc.:Wallingford,CT,2009.
(25) Maseras,F.;Morokuma,K.J.Comput.Chem.1995,16,1170.
(26) Humbel,S.;Siebe,R.S.;Morokuma,K.J.Chem.Phys.1996, 105,1959.
(27) Lesthaeghe,D.;Speybroeck,V.V.;Marin,G.B.;Waroquier,M. Chem.Phys.Lett.2006,417,309.
(28) Sumpter,B.G.J.Chem.Theory Comput.2010,6,727.
(29) Rigby,A.M.;Kramer,G.J.;van Santen,R.A.J.Catal.1997, 170,1.
(30) Zheng,X.;Blowers,P.J.Phys.Chem.A 2006,110,2455.
(31) Zheng,A.M.;Zhang,H.L.;Lu,X.;Liu,S.B.;Deng,F.J.Phys. Chem.B 2008,112,4496.
(32)Zheng,A.M.;Huang,S.;Chen,W.;Wu,P.;Zhang,H.;Lee,H.; Ménorval,L.;Deng,F.;Liu,S.B.J.Phys.Chem.A 2008,112, 7337.
(33) Brand,H.V.;Curtiss,L.A.;Iton,L.E.J.Phys.Chem.1993,97, 12773.
(34) Datka,J.;Boczar,M.;Rymarowicz,P.J.Catal.1988,114,368.
(35) Dunne,J.A.;Rao,M.;Sircar,S.;Corte,R.J.;Myers,A.L. Langmuir 1996,12,5896.
(36) Savitz,S.;Siperstein,F.;Rorte,R.J.;Myers,A.L.J.Phys. Chem.B 1998,102,6865.
(37) Lee,C.;Parrillo,D.J.;Gorte,R.J.;Farneth,W.E.J.Am.Chem. Soc.1996,118,3262.
(38) Fuchs,A.H.;Adamo,C.J.Phys.Chem.Lett.2010,1,763.
October 9,2011;Revised:November 17,2011;Published on Web:November 23,2011.
Adsorption Structure and Energy of Pyridine Confined inside Zeolite Pores
HAN Bing1,2CHU Yue-Ying1,2ZHENGAn-Min1,*DENG Feng1,*
(1Wuhan Center for Magnetic Resonance,State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences,Wuhan 430071,P.R.China;2Graduate University of Chinese Academy of Sciences,Beijing 100049,P.R.China)
The performance of different exchange-correlation functionals for the description of the interaction of pyridine with different cluster models of ZSM-5 zeolite has been assessed.Theoretical calculations show that upon increasing the cluster model from 8T to 128T,the adsorption energy of pyridine in ZSM-5 zeolite increases gradually and reaches convergence by the 72T cluster model.On the basis of the 72T cluster model,the pyridine adsorption energy calculated with different functionals is further examined.Compared to the conventional functionals(B3LYP and M06-2X),the B97D functional which takes into account the dispersion correction provides calculated results that agree well with experimental data.The present results indicate that the B97D functional is suitable for studying long-range interactions in weakly interacting systems.
Quantum chemical calculation;Zeolite;Confinement effect;Adsorption energy
10.3866/PKU.WHXB201111232
*Corresponding authors.ZHENGAn-Min,Email:zhenganm@wipm.ac.cn;Tel:+86-27-87197359.DENG Feng,Email:dengf@wipm.ac.cn;
Tel:+86-27-87198820.
The project was supported by the National Natural Science Foundation of China(21073228,20933009,20921004)and National Key Basic Research Program of China(973)(2009CB918600).
国家自然科学基金(21073228,20933009,20921004)及国家重点基础研究发展规划项目(973)(2009CB918600)资助
O641
