靛红类双功能抑制剂:调控SARS-3CL蛋白酶聚集状态与活性
2012-12-11尚尔昌李春梅来鲁华
周 璐 金 凤 刘 莹,2,* 尚尔昌 魏 平 李春梅 来鲁华,2,*
(1北京大学化学与分子工程学院物理化学研究所,分子动态与稳态结构国家重点实验室,北京分子科学国家实验室,北京100871;2北京大学定量生物学中心,北京100871;3复旦大学药学院,上海201203)
靛红类双功能抑制剂:调控SARS-3CL蛋白酶聚集状态与活性
周 璐1,3金 凤1刘 莹1,2,*尚尔昌1魏 平1李春梅1来鲁华1,2,*
(1北京大学化学与分子工程学院物理化学研究所,分子动态与稳态结构国家重点实验室,北京分子科学国家实验室,北京100871;2北京大学定量生物学中心,北京100871;3复旦大学药学院,上海201203)
1-(2-萘甲基)靛红-5-甲酰胺类化合物通过与底物口袋结合来抑制SARS-3CL蛋白酶的活性,而SARS-3CL蛋白酶自身的N端8肽是作用于蛋白二聚界面的抑制剂.本文设计同时占据SARS-3CL蛋白酶底物口袋和二聚界面的双功能抑制剂,通过固相多肽合成方法制备由1-(2-萘甲基)靛红-5-甲酸和N端8肽组成的化合物,得到不同长度连接链的6个目标产物.用显色底物方法测定化合物对SARS-3CL蛋白酶的抑制活性,其中化合物3的活性最高,IC50值(半抑制率)为3.8 μmol·L-1,连接偶数甘氨酸的活性明显要好于连接奇数甘氨酸的化合物.用超速离心沉降速率方法研究了化合物3对SARS-3CL蛋白酶聚集状态与活性的调控作用,其同时具有诱导与抑制二聚的双重能力,综合调控结果是抑制SARS-3CL蛋白酶的二聚.这项研究给应用合成的化合物研究酶活性调节机制提供了一个示例.
靛红类衍生物;调控;SARS-3CL蛋白酶;二聚;双功能抑制剂
1 Introduction
Isatin(1H-indole-2,3-dione)and its derivatives can be used as the starting material for preparing quinazoline drugs(such as cinchophen).1,2These compounds demonstrate a diverse array of biological and pharmacological activities including anticonvulsant,3antibacterial,4,5antifungal,6antiviral,7-9and anticancer10,11properties.
In 2003,a public infectious disease——severe acute respiratory syndromes(SARS)was outbreak.The pathogen was a new kind of coronavirus12which was named as SARS Coronavirus (SARS-CoV)by World Health Organization(WHO).The main proteinase of SARS-CoV(SARS-3CL proteinase,SARS-3CL) is the key enzyme in the viral life-cycle,which has no homologous proteins in the human cells,with a low mutation rate in virus replication.SARS-3CL was proposed as a key target for drug design against SARS and other diseases caused by coronavirus.13,14
Shortly after the SARS outbreak,several research groups designed SARS-3CL inhibitors from reported inhibitors of human rhinovirus 3C protease(HRV-3C Pro)in order to accelerate the SARS-3CL inhibitor discovery,for both enzymes possessing similarity in their active pocket structure.A set of N-substituted isatin derivatives reported in 1995 by Webber et al.9were confirmed to be SARS-3CL inhibitors.15-17Our previous research16demonstrated that 1-(2-naphthlmethyl)isatin-5-formamide(5f)(Fig.1)is a non-covalent SARS-3CL inhibitor with IC50(half maximal inhibitory concentration).
We have shown that dimerization of SARS-3CL is necessary for enzymatic activity.18,19The dissociation constant of dimerization was determined to be 14.0 μmol·L-1from analytical ultracentrifuge study using purified protein.20However,the apparent dissociation constant measured from concentration dependent enzyme activity study was much lower,indicating a clear tendency of substrate enhanced dimerization.As 5f has a similar chemical structure with the SARS-3CL substrate,it can also induce SARS-3CL dimer formation.21We have used 5f as a probe to study the maturation mechanism of SARS-3CL and proposed a new model.22
The N-terminal octapeptide in SARS-3CL located at the dimer interface.The chemically synthesized octapeptide competes with the SARS-3CL dimer formation23and acts as an allosteric SARS-3CL inhibitor.21In the present study,we linked 5f and the octapeptide together via various linkers and studied their effect on the protein dimerization and enzyme activity.
2 Experimental
2.1 Materials and instruments
The amino acids used for peptide synthesis in this paper were ordered from GL Biochem(Shanghai)Ltd.,Advanced ChemTech Co.USA(>98%).Amino acid with side chain protection:Fmoc-Arg(Pbf),Fmoc-Lys(Boc)Fmoc-Ser(OBut).
PS3™small automatic peptide synthesis apparatus(PTI Co., USA)was used for the synthesis.Micromass GCT mass spectrometer(Manchester,England)was used to measure the molecular weights of the synthesized compounds.High performance liquid chromatography(RP-HPLC,Elite P200II,Dalian,China)system was used for purification.
Others:Zorbax C18 semi-preparative column(9.4 by 250 mm,Agilent).Multiwell ultraviolet spectrometer(Spectra Max 190,Molecular Device,United States).Beckman Optima XLA analytical ultracentrifuge(Canada).
2.2 Computational and synthesis method
Based on the complex crystal structure of SARS-3CL with a small molecule inhibitor(ethyl-(2E,4S)-4-{[(2S)-2-{[N-(tertbutoxycarbonyl)-L-valyl]amino}-2-phenylacetyl]amino}-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate)(PDB code:2d2d),24we analyzed the SARS-3CL dimer interface and calculated the distance between the interfacial inhibitor(N-terminal octapeptide)and active site inhibitor(5f).The two inhibitors were linked with a polyglycine chain with suitable length.In order to form the new amide bond between 5f and N-terminal octapeptide,1-(2-naphthlmethyl)isatin-5-formic acid(5fʹ)(Fig.1) was used as a starting material,through the dehydration of OH groups at C-5 of 5fʹand H at N1 of amino acid.
Solid-phase peptide synthesis method was used to prepare the following title compounds(Fig.1).
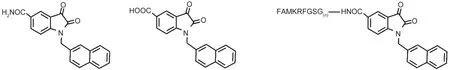
Fig.1 Chemical structures of the inhibitors 5f(left),5fʹ(middle),and dual functional inhibitors(right)
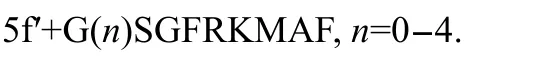
Briefly,the small molecule modified peptides were synthesized on rink resin using standard Fmoc/DIC/HOBt chemistry. The deprotection of the Fmoc and t-Bu groups was treated with reagent R(90%trifluoro acetic acid(TFA),5%thioanisole,5%water).The products were purified by high performance liquid chromatography(HPLC)and characterized by electronicspray ionization mass spectra(ESI-MS)before determination of the biological activities.
2.3 Continuous colorimetric assay
The inhibition activity was measured by continuous colorimetric assay25using colorimetric substrate Thr-Ser-Ala-Val-Leu-Gln-pNA(HPLC purity).SARS-3CL was cloned and expressed in Escherichia coli and purified according to the published procedure.21Before the inhibition test,the enzyme buffer was changed to phosphate buffered saline(PBS)(20 mmol·L-1, pH 7.4).Colorimetric measurements of the protease activity were performed in 96-well microtiter plates using a multiwell ultraviolet spectrometer.Substrate(final concentration of 200 μmol·L-1)and inhibitors were mixed together in 20 mmol·L-1PBS buffer(pH 7.4)in the presence of 5%(V/V)dimethyl sulfoxide,and the reaction was initiated by adding SARS-3CL(final concentration of 1 μmol·L-1).
2.4 Sedimentation velocity experiments
The sedimentation experiments can be used to study the aggregation state of proteins in the native solution condition,and no dilution effect exists as compared to gel filtration which may cause inhibitors deposition.Sedimentation velocity provides hydrodynamics information about the molecular size distribution and conformational changes.We used sedimentation velocity experiments to determine the dimerization of SARS-3CL when inhibitors were presented.
Our sedimentation velocity experiments were conducted on a Beckman Optima XLA analytical ultracentrifuge equipped with absorbance optics.21Data were analyzed with the software Sedfit version Sedfit v9.4(http://www.analyticalul-tracentrifugation.com/).Different inhibitors were tested to examine whether they can disturb the enzyme dimerization.The concentrations of enzyme and inhibitors were 9 and 10 μmol·L-1,respectively.The buffer contained 100 mmol·L-1NaCl,20 mmol·L-1NaH2PO4-Na2HPO4(pH=7.4),and 0.5%(V/V)dimethylsulfoxide(DMSO).
3 Results and discussion
3.1 Design and synthesis of dual function inhibitors Isatin inhibitors of HRV-3C Pro9were docked into the substrate binding pocket of SARS-3CL using the SYBYL program (Tripos,Inc.),in a similar orientation as that of the covalent binding substrate analogue in the published complex crystal structure.24The carbonyl oxygen atoms at C-2 and C-3 have hydrogen bonds with the active site residues His41 and Cys145. The carboxamide at C-5 holds the same position as that of the substrate P1 Gln side chain through H-bonds with the backbone carbonyl of Phe140 and the imidazole ring of His163.Alkyl groups at N-1 fit into the hydrophobic S2 site formed by Met49 and Met165.16
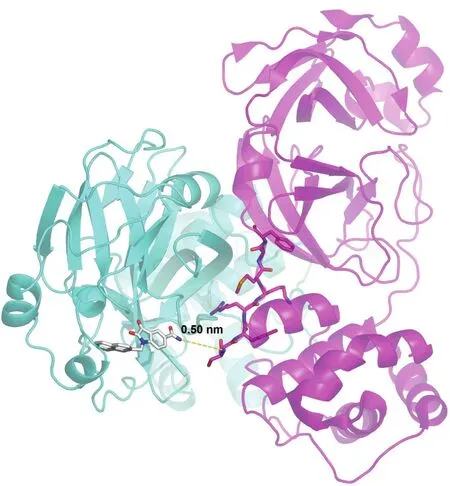
Fig.2 Distance between active site inhibitor 5f and N-terminal octapeptide in the other monomer of the SARS-3CLdimer
N-terminal octapeptide of SARS-3CL locates at the dimer interface and itself was reported to act as an interfacial inhibitor of SARS-3CL since SARS-3CL existed as a mixture of dimer and monomer in solution and only the dimer was considered to be active.18,19Three residues of the N-terminal octapeptide play key roles in the binding model:Met6 in the N-terminal octapeptide is accommodated in a hydrophobic pocket;Ser1 and the side chain ofArg4 fit in two negative charge pockets.20
Supposing that the individual N-terminal octapeptide interacted with one protease monomer in the same way as it is in the other monomer,the distance between serine at N1 of the octapeptide and C5 carboxamide isatin inhibitors at active site was quite short(about 0.50 nm,Fig.2).
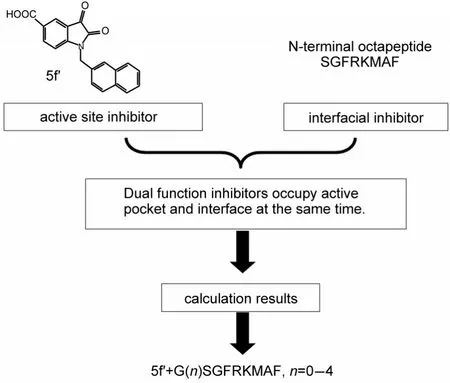
Fig.3 Design strategy for SARS-3CLdual functional inhibitor
Based on the model structure,we designed the dual functional inhibitor by linking the two inhibitors through a suitable linker(Fig.3).We hope the dual functional inhibitor only bind to the protein monomer,thus disturbing the dimerization of SARS-3CL which makes it bifunctional.For synthesis convenience,we used glycine as linker unit because glycine is the most flexible residue among all the amino acids.We screened linker length from 0 to 4 glycine residues since the distance between two linking sites is 0.50 nm which is about two glycine residues.Mutations at the octapeptide or deletion of the whole octapeptide were also designed to examine the interface binding effect of the octapeptide.
The new compounds with 5f linking SGFRKMAF or its mutants were synthesized by standard Fmoc solid-phase peptide synthesis method.The structure information of these compounds is listed in Table 1.
3.2 Enzyme inhibition assay
For all compounds,we tested their inhibition against SARS-3CL.The IC50values are listed in Table 1.
Among all the dual functional compounds,compound 3 showed the strongest inhibition,with an IC50of(3.8±0.2) μmol·L-1.It was necessary to link 5f and the octapeptide by two glycine residues to keep both parts at a suitable binding conformation,which was consistent with the computational analysis.Compound 1 showed no activity at 100 μmol·L-1due to missing connection linkers.Compounds 2-5 are all active with the length of the polyglycine chains of 1 to 4.Inhibitors with the even spacers showed better activity than the odd ones, which could be explained by the angle restriction of peptide bonds.In order to verify the binding of the octapeptide to the protein,compound 7 with only 5fʹlinked to two glycines was synthesized.Compound 7 has weak inhibition activity compared to compounds 2-5,indicating that the N-terminal octapeptide is involved in protein binding.We also synthesized compounds with mutations in the octapeptide and measured their inhibition activity.For example,compound 6 contains two mutations(F3T and F8T)in the octapeptide to destroy the hydrophobic interactions,was found to be less active.On the other hand,the inhibition of compounds 2-5 is lower than that of compound 5f,which implies that the dual functional inhibitors may have different mechanism.
3.3 Analytical ultracentrifugation analysis
Sedimentation velocity experiments can analyze the aggregation distribution of macromolecules in solution sensitively.Weused analytical ultracentrifugation to study SARS-3CL dimer formation incubated with different concentrations of compounds 3 and 5f(Fig.4).As shown before,compound 5f dramatically induced dimerization of SARS-3CL16while binding to the substrate binding site as a competitive inhibitor.When 5f was linked to the N-terminal octapeptide by two glycines(compound 3),the dimer/monomer ratio dramatically reduced compared to that of 5f.This indicates that the octapeptide can still prevent SARS-3CL dimer formation when linked to 5f.Thus in the dual functional inhibitor compound 3,the 5f’part inhibits the SARS-3CL activity by binding to the substrate binding site,and the octapeptide head prevents the SARS-3CL dimer formation.
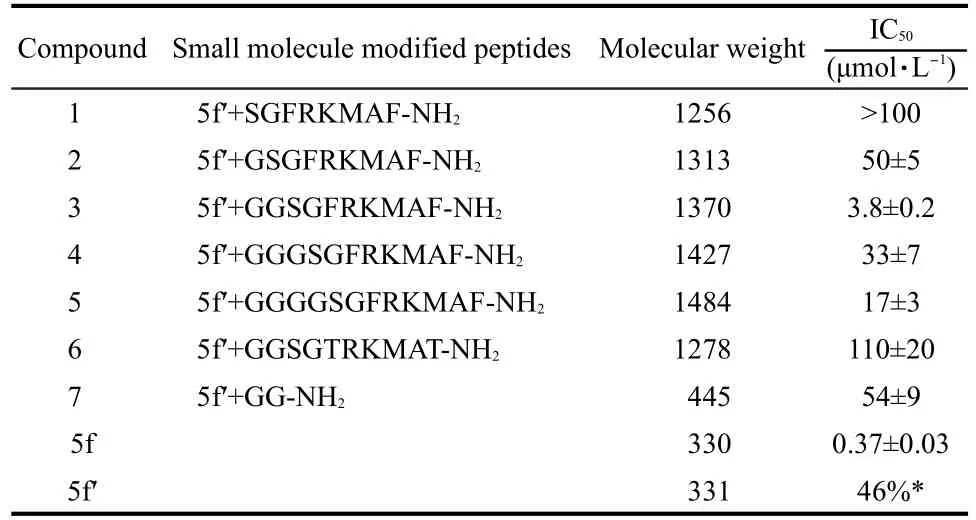
Table 1 Compound structures and activity data
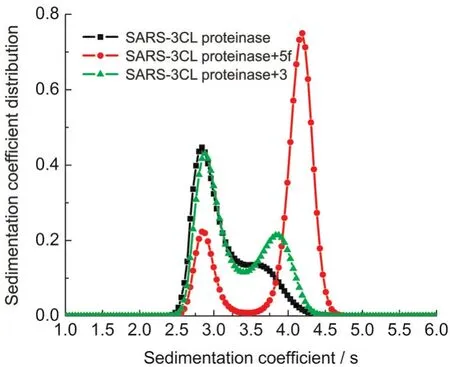
Fig.4 Effects of inhibitors on the dimerization of SARS-3CL
4 Conclusions
We have synthesized a series of dual functional inhibitors for SARS-3CL by linking the N-terminal octapeptide and an active site isatin inhibitor using a polyglycine linker.The change of inhibition activity with the linker length was studied. The optimum linker length was found to be two glycine residues,and the corresponding compound 3 showed the highest inhibition activity with an IC50of 3.8 μmol·L-1.Sedimentation velocity experiments showed that the dual functional inhibitor (compound 3)induced less SARS-3CL dimer formation compared to 5f.This indicates that when designing dual functional inhibitors,both the active site binding affinity and the dimer induction ability needs to be considered.Compared to the original N-terminal octapeptide(IC50=0.8 mmol·L-1),the inhibition activity of compound 3 is dramatically increased.However, due the block of the amide group in 5f,the activity of compound 3 is lower than that of 5f(IC50=0.37 μmol·L-1).As 5f is a universal inhibitor for both 3C-like and 3C proteases in various viruses,attaching the SARS-3CL specific N-terminal octapeptide may increase its selectivity.This work provides an example of using synthesized compounds to study enzyme activity regulation mechanism.
(1) Bogert,M.T.;Nabenhauer,F.P.J.Am.Chem.Soc.1924,46, 1702.doi:10.1021/ja01672a021
(2) Trinka,P.;Slegel,P.;Reiter,J.J.Prakt.Chem.1996,338,675. doi:10.1002/(ISSN)1521-3897
(3)Praveen,C.;Ayyanar,A.;Perumal,P.T.Bioorg.Med.Chem. Lett.2011,21,4072.doi:10.1016/j.bmcl.2011.04.117
(4)Shingade,S.G.;Bari,S.B.;Waghmare,U.B.Med.Chem.Res. 2012,21,1302.doi:10.1007/s00044-011-9644-y
(5)Adibi,H.;Khodaei,M.;Pakravan,P.;Abiri,R.Pharm.Chem.J. 2010,44,219.doi:10.1007/s11094-010-0436-3
(6) Pervez,H.;Iqbal,M.S.;Tahir,M.Y.;Nasim,F.U.;Choudhary, M.I.;Khan,K.M.J.Enzym.Inhib.Med.Chem.2008,23,848. doi:10.1080/14756360701746179
(7) Pervez,H.;Lqbal,M.S.;Tahir,M.Y.;Choudhary,M.I.;Khan, K.M.Nat.Prod.Res.2007,21,1178.doi:10.1080/ 14786410601129770
(8) Dragovich,P.S.;Prins,T.J.;Zhou,R.;Webber,S.E.; Marakovits,J.T.;Fuhrman,S.A.;Patick,A.K.;Matthews,D. A.;Lee,C.A.;Ford,C.E.;Burke,B.J.;Rejto,P.A.; Hendrickson,T.F.;Brown,E.L.;Meador,J.W.;Ferre,R.A.; Harr,J.E.V.;Kosa,M.B.;Worland,S.T.J.Med.Chem.1999, 42,1213.doi:10.1021/jm9805384
(9) Webber,S.E.;Tikhe,J.;Worland,S.T.;Fuhrman,S.A.; Hendrickson,T.F.;Matthews,D.A.;Love,R.A.;Patick,A.K.; Meador,J.W.;Ferre,R.A.;Brown,E.L.;DeLisle,D.M.;Ford, C.E.;Binford,S.L.J.Med.Chem.1996,39,5072.doi:10.1021/ jm960603e
(10) Vine,K.L.;Matesic,L.;Locke,J.M.;Ranson,M.;Skropeta,D. Anti-Cancer Agents Med.Chem.2009,9,397.
(11)Kandile,N.G.;Mohamed,M.I.;Ismaeel,H.M.J.Enzym. Inhib.Med.Chem.2012,27,330.doi:10.3109/ 14756366.2011.588950
(12) Peiris,J.S.M.;Lai,S.T.;Poon,L.L.M.;Guan,Y.;Yam,L.Y. C.;Lim,W.;Nicholls,J.;Yee,W.K.S.;Yan,W.W.;Cheung, M.T.;Cheng,V.C.C.;Chan,K.H.;Tsang,D.N.C.;Yung,R. W.H.;Ng,T.K.;Yuen,K.Y.Lancet 2003,361,1319.doi: 10.1016/S0140-6736(03)13077-2
(13)Yang,H.T.;Yang,M.J.;Ding,Y.;Liu,Y.W.;Lou,Z.Y.;Zhou, Z.;Sun,L.;Mo,L.J.;Ye,S.;Pang,H.;Gao,G.F.;Anand,K.; Bartlam,M.;Hilgenfeld,R.;Rao,Z.H.Proc.Natl.Acad.Sci. U.S.A.2003,100,13190.doi:10.1073/pnas.1835675100
(14) Lai,L.H.;Han,X.F.;Chen,H.;Wei,P.;Huang,C.K.;Liu,S. Y.;Fan,K.Q.;Zhou,L.;Liu,Z.M.;Pei,J.F.;Liu,Y.Curr. Pharm.Des.2006,12,4555.doi:10.2174/ 138161206779010396
(15)Chen,L.R.;Wang,Y.C.;Lin,Y.W.;Chou,S.Y.;Chen,S.F.; Liu,L.T.;Wu,Y.T.;Chih-Jung,K.B.;Chen,T.S.S.;Juang,S. H.Bioorg.Med.Chem.Lett.2005,15,3058.doi:10.1016/ j.bmcl.2005.04.027
(16) Zhou,L.;Liu,Y.;Zhang,W.L.;Wei,P.;Huang,C.K.;Pei,J.F.; Yuan,Y.X.;Lai,L.H.J.Med.Chem.2006,49,3440.doi: 10.1021/jm0602357
(17) Liu,Y.;Zheng,T.F.;Jin,F.;Zhou,L.;Liu,Z.M.;Wei,P.;Lai, L.H.Acta Chim.Sin.2007,65,1707.[刘 莹,郑腾飞,金 凤,周 璐,刘振明,魏 平,来鲁华.化学学报,2007,65, 1707.]
(18)Fan,K.Q.;Wei,P.;Feng,Q.;Chen,S.D.;Huang,C.K.;Ma, L.;Lai,B.;Pei,J.F.;Liu,Y.;Chen,J.G.;Lai,L.H.J.Biol. Chem.2004,279,1637.doi:10.1074/jbc.M310875200
(19) Chen,H.;Wei,P.;Huang,C.K;Tan,L.;Liu,Y.;Lai,L.H. J.Biol.Chem.2006,281,13894.doi:10.1074/jbc.M510745200
(20) Wei,P.;Fan,K.Q.;Chen,H.;Ma,L.;Huang,C.K.;Tan,L.;Xi, D.;Li,C.M.;Liu,Y.;Cao,A.N.;Lai,L.H.Biochem.Biophys. Res.Commun.2006,339,865.doi:10.1016/j.bbrc.2005.11.102
(21) Wei,P.;Li,C.M.;Zhou,L.;Liu,Y.;Lai,L.H.Acta Phys.-Chim. Sin.2010,26,1093.[魏 平,李春梅,周 璐,刘 莹,来鲁华.物理化学学报,2010,26,1093.]doi:10.3866/PKU.WHXB 20100449
(22)Li,C.M.;Qi,Y.F.;Teng,X.;Yang,Z.C.;Wei,P.;Zhang,C.S.; Tan,L.;Zhou,L.;Liu,Y.;Lai,L.H.J.Biol.Chem.2010,285, 28134.doi:10.1074/jbc.M109.095851
(23) Fan,K.Q.;Ma,L.;Han,X.F.;Liang,H.H.;Wei,P.;Liu,Y.; Lai,L.H.Biochem.Biophys.Res.Commun.2005,329,934. doi:10.1016/j.bbrc.2005.02.061
(24)Yang,H.T.;Xie,W.Q.;Xue,X.Y.;Yang,K.L.;Ma,J.;Liang, W.X.;Zhao,Q.;Zhou,Z.;Pei,D.Q.;Ziebuhr,J.;Hilgenfeld, R.;Yuen,K.Y.;Wong,L.;Gao,G.X.;Chen,S.J.;Chen,Z.; Ma,D.W.;Bartlam,M.;Rao,Z.H.PLoS Biol.2005,3,1742.
(25) Huang,C.K.;Wei,P.;Fan,K.Q.;Liu,Y.;Lai,L.H. Biochemistry 2004,43,4568.doi:10.1021/bi036022q
July 25,2012;Revised:September 10,2012;Published on Web:September 14,2012.
Isatin Dual Functional Inhibitors:Modulating the Aggregation State and Enzyme Activity of SARS-3CL Proteinase
ZHOU Lu1,3JIN Feng1LIU Ying1,2,*SHANG Er-Chang1WEI Ping1LI Chun-Mei1LAI Lu-Hua1,2,*
(1Beijing National Laboratory for Molecular Sciences,State Key Laboratory of Structural Chemistry of Unstable and Stable Species, Institute of Physical Chemistry,College of Chemistry and Molecular Engineering,Peking University,Beijing 100871,P.R.China;2Center for Quantitative Biology,Peking University,Beijing 100871,P.R.China;3School of Pharmacy,Fudan University,Shanghai 201203,P.R.China)
The 1-(2-naphthlmethyl)isatin-5-formamide compounds can inhibit SARS-3CL proteinase by binding to its substrate pocket,while the N-terminal octapeptide of SARS-3CL proteinase was found to act as a dimerization inhibitor.In this work,the dual functional inhibitors which can occupy both substrate pocket of SARS-3CL proteinase and its dimer interface were designed.Six title compounds were gotten by linking 1-(2-naphthlmethyl)isatin-5-formic acid and N-terminal octapeptides using a polyglycine linker through solid-phase peptide synthesis method.The in vitro inhibition activity against SARS-3CL proteinase was measured by continuous colorimetric assay using colorimetric substrate.Compound 3 showed the highest inhibition activity with an IC50(half maximal inhibitory concentration of a substance)of 3.8 μmol·L-1. The change of inhibition activity with the linker length was studied.Inhibitors with the even spacers were showed better activity than the odd ones,which could be explained by the angle restriction of peptide bonds.The modulating of the aggregation state and enzyme activity towards SARS-3CL proteinase were studied using sedimentation velocity experiments.Compound 3 was found to not only inhibit the enzyme activity of SARS-3CL proteinase,but also shift the monomer-dimer equilibrium of the enzyme.The integrated control result is inhibiting SARS-3CL proteinase dimer formation.This work provides an example of using synthesized compounds to study enzyme activity regulation mechanism.
Isatin derivative;Modulation;SARS-3CL proteinase;Dimerization;Dual functional inhibitor
10.3866/PKU.WHXB201209143
∗Corresponding authors.LIU Ying,Email:liuying@pku.edu.cn;Tel:+86-10-62751490.LAI Lu-Hua,Email:lhlai@pku.edu.cn; Tel:+86-10-62757486.
The project was supported by the National Natural Science Foundation of China(90913021,20473001,11021463)and National Key Basic Research Program of China(973)(2009CB9185003).
国家自然科学基金(90913021,20473001,11021463)和国家重点基础研究发展规划项目(973)(2009CB9185003)资助
O641
