微介孔复合分子筛合成及其在汽车冷启动尾气控制中的应用
2012-12-11张振中尉继英孟弼芳梁彤祥
张 兰 张振中 尉继英 孟弼芳 江 锋 梁彤祥
(清华大学核能与新能源技术研究院,精细陶瓷北京市重点实验室,北京100084)
微介孔复合分子筛合成及其在汽车冷启动尾气控制中的应用
张 兰 张振中 尉继英*孟弼芳 江 锋 梁彤祥
(清华大学核能与新能源技术研究院,精细陶瓷北京市重点实验室,北京100084)
分别采用稀溶液和浓溶液双模板剂两步水热合成法,以二氧化硅微硅粉和铝酸钠为硅源和铝源,第一步获得β分子筛晶种,第二步以β分子筛晶种为结构单元组装形成兼具MCM-41分子筛和β分子筛结构特点的复合型分子筛β/M(其中β是指β分子筛,M是指MCM-41介孔分子筛).采用X射线衍射(XRD)、傅里叶变换红外(FT-IR)光谱、氮气吸附(BET)和高分辨透射电镜(HRTEM)对样品的物相结构和微观形貌进行了表征,并对复合分子筛的合成过程进行了分析.结果表明:复合分子筛β/M的形成是微孔β结构和介孔MCM-41结构的竞争生长过程,β分子筛晶种的晶化时间和晶粒度大小对β/M复合分子筛的结构有重要影响.此外,我们以甲苯为探针分子,比较研究了三类分子筛β/M、MCM-41和β的原样以及经高温水热处理后样品的吸附性能,结果表明:β/M复合分子筛的热稳定性优于介孔分子筛MCM-41,其甲苯吸附容量比MCM-41和β分子筛的高,其中以浓溶液法合成的复合分子筛吸附容量最高.
微介孔复合分子筛;晶化时间;晶粒度;甲苯吸附;汽车冷启动尾气控制
1 引言
在汽车冷启动的前2 min由于安装的三元催化器未达到起燃温度,大量燃烧不完全产生的碳氢化合物(HCs)直接排放,从而产生了冷启动问题,使汽车无法达到更高的排放标准.对冷启动尾气污染物的净化是现阶段的重要任务.HC-trap技术是最有发展前景的冷启动尾气净化技术之一.1,2该技术采用分子筛在低温下吸附HCs,当三元催化器起燃后,解析出来的HCs经过三元催化器转变为CO2和H2O.该技术要求分子筛具有良好的高温水热稳定性,对芳香烃大分子的吸附容量高,而且对短链小分子HCs的解析温度不低于200°C.3,4微孔分子筛β是目前吸附性能最好的HC-trap材料,5但孔体积小制约其吸附容量;常规的介孔分子筛孔体积大,但因其孔壁为无定形结构,其酸性、水热稳定性较差,难以应用于冷启动尾气吸附.
美国密歇根州立大学的Pinnavaia等、6-10中国吉林大学的肖丰收等、11-13比利时安特卫普大学的Van Oers等14以及中国科学院太原煤炭化学研究所的郑均林等15,16尝试将ZSM-5、β等微孔分子筛的初级或次级结构引入介孔分子筛的孔壁结构中,形成微介孔复合分子筛,保留了微孔分子筛的酸性以及介孔分子筛较大的比表面积和孔容,同时还具有较好的水热稳定性,有着广阔的应用前景.17-19这为HCs-Trap技术提供了新的探索思路.
本文采用两步水热合成法,以微孔分子筛β的晶种为结构单元,通过β晶种自组装构筑新型孔壁晶化的介孔分子筛,并以甲苯为探针分子,研究该复合分子筛对芳香烃大分子的吸附性能.
2 实验
2.1 试 剂
铝酸钠(NaAlO2,AR),二氧化硅微硅粉(SiO2, AR),氢氧化钠(NaOH,AR),四乙基氢氧化铵(TEAOH,AR,20%水溶液),十六烷基三甲基溴化铵(CTAB,AR),以上试剂均出自北京国药试剂厂;超纯水(H2O,电阻率为18.2 MΩ·cm).
2.2 样品制备
2.2.1 MCM-41分子筛的制备
以NaAlO2、SiO2、NaOH、CTAB和H2O为原料,制备Si/Al摩尔比为90的MCM-41,具体制备过程见文献.20
2.2.2 β分子筛的制备
以NaAlO2、SiO2、NaOH、TEAOH和H2O为原料,其各物质摩尔比为1:30:0.06:0.3:20,在室温下搅拌一定的时间后,形成均匀的胶体溶液,后转移至聚四氟乙烯反应釜中,在140°C条件下晶化72 h,得到β分子筛原粉,洗涤,烘干,并在550°C下焙烧6 h,得到β分子筛.
2.2.3 复合分子筛的制备
复合分子筛的制备采用两步水热合成法:第一步是β晶种的制备,第二步是β晶种在CTAB模板剂作用下自组装成为复合分子筛样品.操作过程如下:(1)将NaAlO2、SiO2、NaOH、TEAOH和H2O以一定的比例混合,其中Si/Al摩尔比为30,水硅摩尔比为9.8,在室温下搅拌一定的时间后,形成均匀的胶体溶液,后转移至聚四氟乙烯反应釜中,在140°C条件下分别晶化8、10、12和16 h,得到β晶种β8、β10、β12和β16,记晶种晶化时间为t.(2)将β晶种滴加至10%左右的CTAB溶液中,并调整溶液pH值到10左右,搅拌均匀后转移至反应釜中,在140°C下晶化48 h,得到β/M分子筛原粉,用去离子水洗涤至中性,然后在80°C下烘干过夜,最后在550°C下焙烧6 h,得到复合型分子筛β/M.根据第一阶段β晶种的晶化时间,将β/M标记为m8、m10、m12和m16.
为了方便表述,把上述晶种制备中水硅摩尔比为9.8的制备方法记为浓溶液合成法.按上述制备步骤,将第一步晶种制备过程的水硅摩尔比调整为20,晶种反应时间设定为20、30、40和50 h,分别得到晶种β20、β30、β40和β50,经第二步组装后,得到复合分子筛样品M20、M30、M40和M50,在该水硅摩尔比下的制备方法记为稀溶液合成法.
2.3 复合分子筛的表征
采用日本理学D/max-RB型X射线衍射仪,Cu靶,测定样品的XRD谱图,低角区扫描范围:2θ= 0.6°-10°,高角区扫描范围:2θ=10°-40°;采用Perkin Elmer Spectrum GX型傅里叶变换红外光谱仪摄取样品的红外谱图,波数范围400-4000 cm-1;采用JEM-2010型高分辨透射电子显微镜观察样品微观形貌;采用ASAP2010比表面分析仪测量样品的比表面积和孔径分布,其中微孔比表面积和孔径分布分别用t-plot法和Saito-Foley(SF)法分析,介孔孔径分布情况用BJH法分析;采用GC112A气相色谱测定分子筛对甲苯的吸附穿透曲线.
3 结果与讨论
3.1 XRD表征
图1为浓溶液和稀溶液合成法制备的复合分子筛样品β/M的XRD谱图.从图中可以看出,在小角衍射区域,所有样品在2θ=2°左右均有较强的衍射峰,对应着六方晶体的(100)晶面,在2θ=3°-7°之间有三个较弱的衍射峰,分别对应六方晶体的(110)、(200)和(210)晶面,说明复合分子筛β/M具有类似于MCM-41分子筛的良好的六方晶相结构.在大角衍射区域,在晶种晶化时间较短时,样品m8、M20、M30大角衍射峰为一谷包,没有显著的衍射峰,这说明此时复合分子筛为单一的物相,21没有形成微孔结构的分子筛;当晶种晶化时间延长,样品m10在2θ=21.5°和2θ=22.5°出现了微弱的衍射峰,分别归属为β分子筛(205)和(116)晶面的特征衍射峰,22说明此时的样品中同时存在介孔相和少量的β微孔相,不再是单一的物相;随着晶种晶化时间继续延长,样品m12、m16以及M40、M50的β特征衍射峰越来越强,并在小角衍射区域2θ=7.8°出现了β分子筛(101)晶面的衍射峰,说明β分子筛晶粒越来越大,介孔相和β微孔相在该复合分子筛中同时存在.
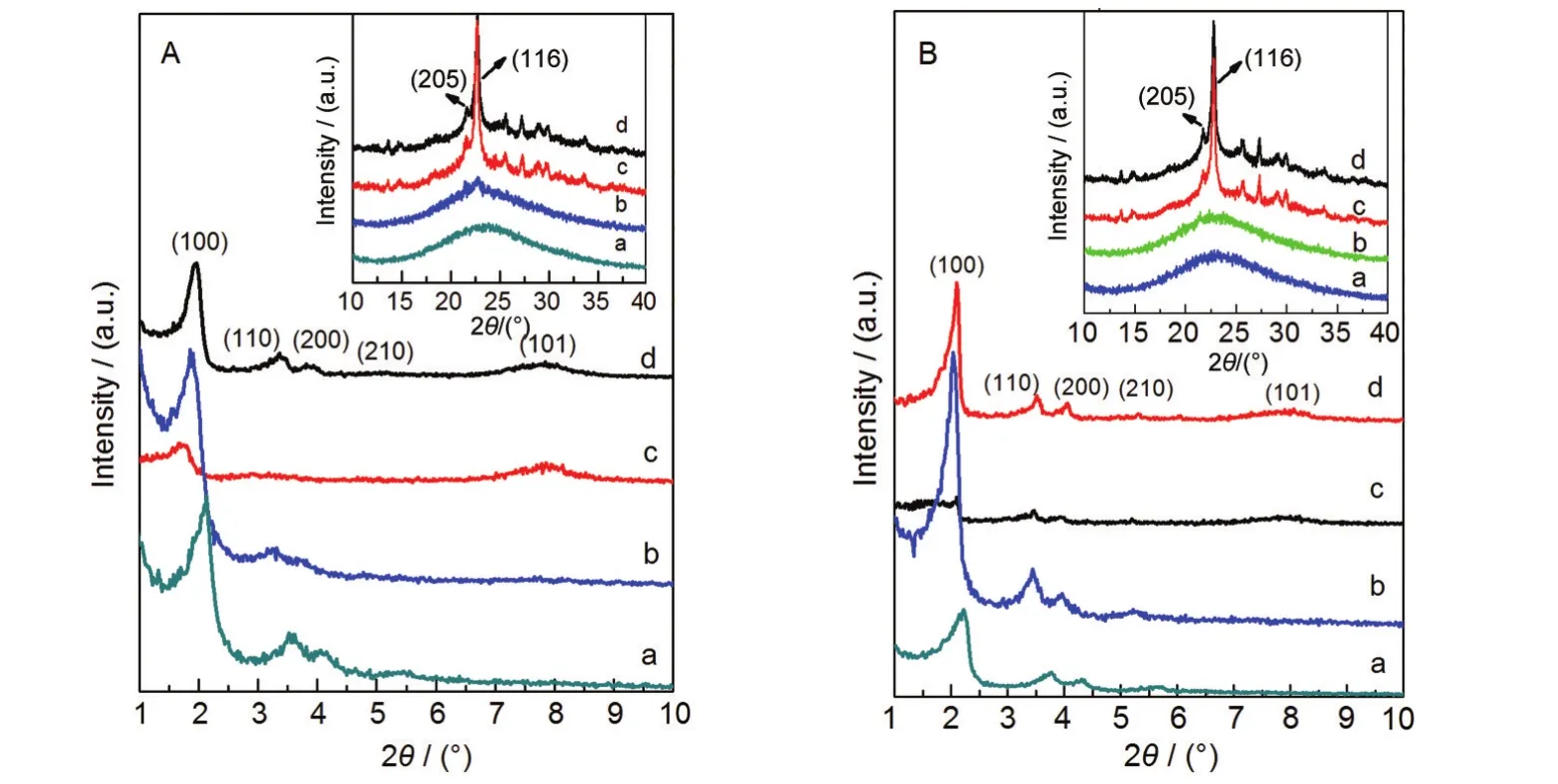
图1 不同晶种晶化时间下制备的复合分子筛样品的XRD图谱Fig.1 XRD patterns of samples with their crystal seed prepared for different time(A)the samples prepared at H2O/SiO2molar ratio of 9.8 in β seeds crystallized for 8,10,12,16 h:(a)m8,(b)m10,(c)m12,(d)m16; (B)the samples prepared at H2O/SiO2molar ratio of 20 in β seeds crystallized for 20,30,40,50 h:(a)M20,(b)M30,(c)M40,(d)M50
从8种复合分子筛样品的XRD衍射结果可以看出:随着β晶种晶化时间的增长,复合分子筛中微孔β晶相越来越明显,物相结构也逐渐由单一介孔变成微孔相和介孔相并存的情形.根据XRD结果,我们认定m8、M20和M30为纯物相,记为βM物相, m12、m16、M40和M50为多物相混合物,m10为βM物相向多物相混合物的过渡阶段.
图2给出了样品m10和MCM-41分别经过高温老化(1073 K焙烧5 h)和高温水热老化(1073 K,通50%水蒸气与50%空气的混合气体)后样品的XRD谱图.从图上可以看出这两种样品在经过高温和水热老化后,其小角衍射区域的特征峰位置均向高角度移动,水热老化样品的位移比高温处理样品大很多.位移的原因可能是在高温及水热处理后分子筛脱铝,导致孔道塌陷,使得晶面间距降低;高温水热处理比高温处理的脱铝现象更加明显,因而晶面间距减少得更多.
通过对比两种样品的XRD谱图发现,经过β晶种介孔组装后的样品m10的特征峰的峰高、峰位移均较MCM-41小很多,也就是说,在高温和水热处理条件下,m10的脱铝程度较MCM-41低,m10具有比MCM-41更高的高温稳定性和水热稳定性.
3.2 FT-IR表征
稀溶液合成法和浓溶液合成法制备的复合分子筛样品的FT-IR谱图如图3所示,我们重点研究了复合分子筛在双环特征波段500-650 cm-1的红外谱带变化情况.当β晶种晶化时间较短时,m8、m10、M20和M30样品在570 cm-1附近出现红外吸收峰,该峰与β五元环振动有关,10m8和M20在该处的吸收峰较宽,而m10和M30的吸收峰较为尖锐.随着样品的晶化时间增加,该吸收峰强度逐渐增强.与m8、M20和M30不同的是,m10在520 cm-1附近出现极其微弱的吸收峰,归属于β四元环振动吸收峰,该峰被认为是β分子筛的特征吸收峰.23这表明m8、M20和M30样品中没有完整的β晶体结构单元存在,只有β次级结构单元——β五元环的存在,而m10样品中已有少量的β完整晶体结构单元存在.样品m12、m16、M40以及M50在524、575和619 cm-1处均出现了尖锐的吸收峰,其中524和619 cm-1处的峰与β四元环振动有关,24575 cm-1处的峰归属于β五元环振动吸收峰,FT-IR结果说明,在晶化时间较长的样品中存在大量的β晶体结构单元.
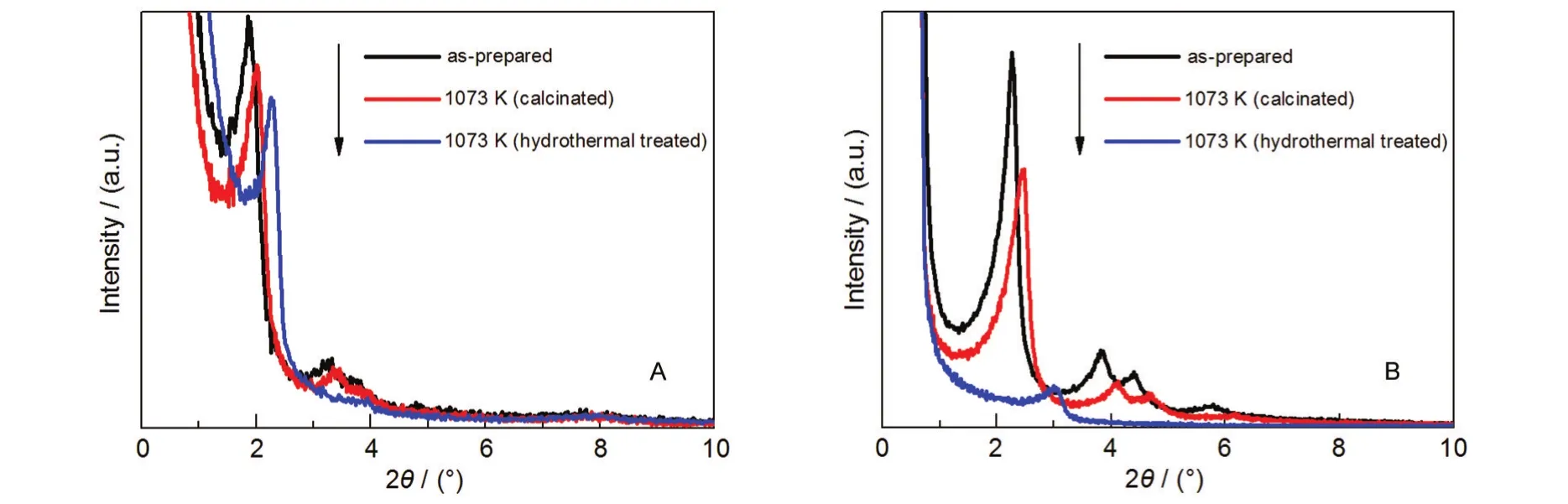
图2 样品m10和MCM-41老化处理前后XRD图谱Fig.2 XRD patterns of m10 and MCM-41 samples before and after aging treatment(A)m10;(B)MCM-41
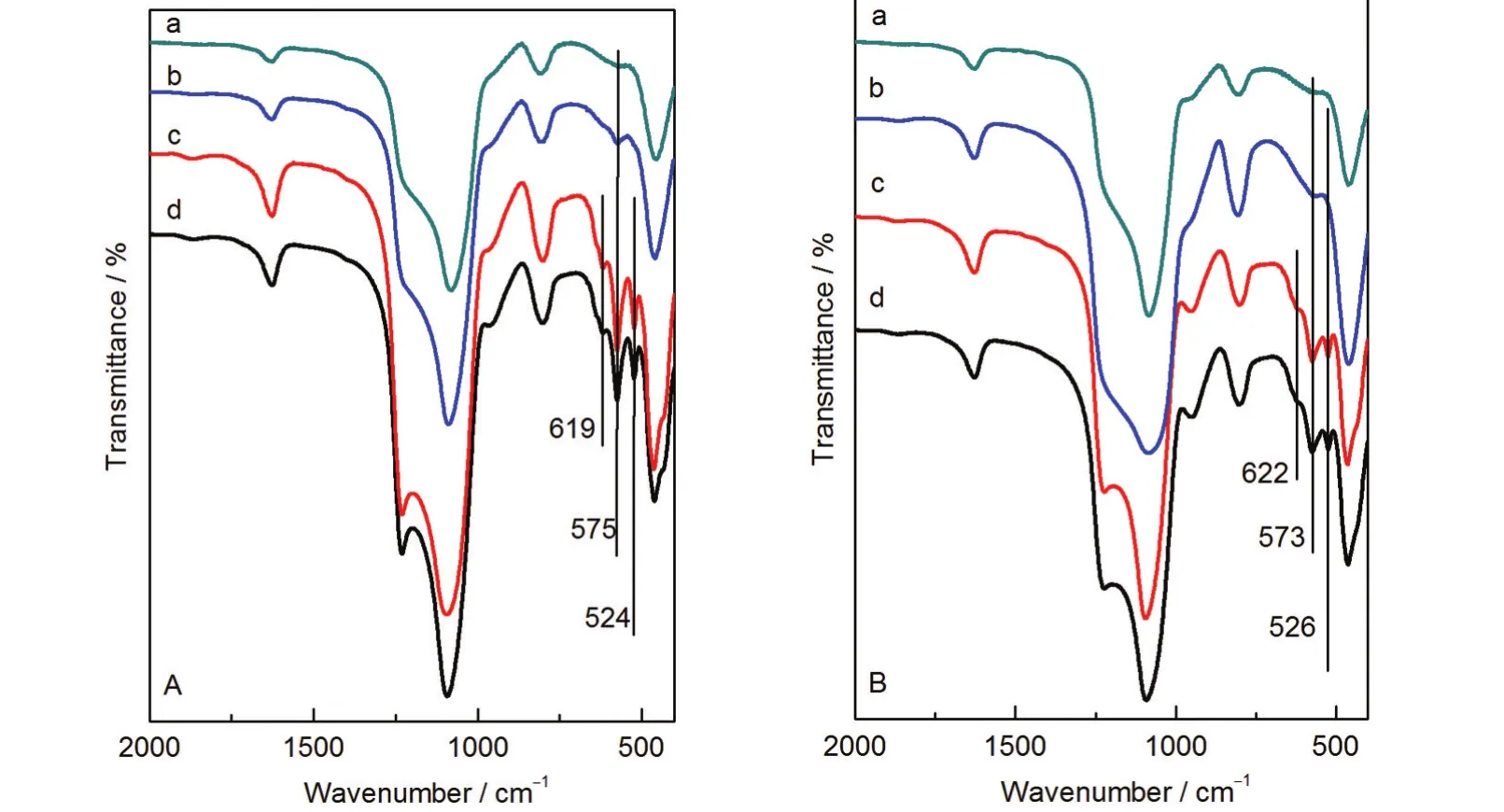
图3 复合分子筛样品的FT-IR图谱Fig.3 FT-IR spectra of combined zeolite samples(A)the samples prepared at H2O/SiO2molar ratio of 9.8:(a)m8,(b)m10,(c)m12,(d)m16; (B)the samples prepared at H2O/SiO2molar ratio of 20:(a)M20,(b)M30,(c)M40,(d)M50
比较不同样品的红外谱图不难发现:随着β晶种的晶化时间延长,在制备的复合分子筛样品中,先是出现β五元环结构的红外吸收峰,并逐渐增强;然后出现β的红外特征吸收峰,并越来越显著;最终出现完整的β分子筛晶体结构的红外吸收峰.这和样品的XRD结果相符,也和纯相β分子筛的晶化过程25一致.这说明最终样品的组成和β晶种晶化时间密切相关,即β晶种的结构和尺寸直接决定着最终样品的物相和结构.
我们推测,当β晶种晶化时间较短时,形成的晶种主要是β五元环结构,且晶粒尺寸较小,容易和硅铝溶胶一起在CTAB模板剂作用下参加介孔孔壁的组装,合成了纯相的βM复合分子筛,母液中存在化学平衡:硅铝溶胶↔β五元环结构↔βM;当晶种晶化时间延长,β晶种中不仅含有大量的五元环结构,并且出现了少量的四元环结构单元,绝大部分晶种颗粒尺寸不大,在CTAB模板剂的作用下有如下化学平衡:β晶体结构↔β五元环结构+硅铝溶胶↔βM,小颗粒的晶粒溶解,体系朝着βM生成的方向进行;当晶种晶化时间更长时,β晶种中有大量的五元环、四元环和六元环结构单元,晶种颗粒的尺寸也逐渐增大,并出现了较大的完整β晶粒,部分尺寸较小的晶种较快溶解,并参加了介孔组装,而尺寸较大的β晶粒由于溶解缓慢,则留在母液中,最终形成了β和βM物相并存的复合分子筛.
结合XRD和FT-IR结果不难得出这样的结果: m8、M20和M30样品为纯相的介孔六方晶体结构,且样品中含有五元环的β次级结构单元,因而我们认为β的五元环次级结构单元参与了介孔分子筛的组装,并已成功引入到介孔分子筛的孔壁之中,是新型的复合分子筛βM;而m12、m16和M40以及M50,由于出现显著的β完整结构的表征结果,我们认为它们是多相混合物,包括微孔β相和βM相;m10则是纯相的βM复合分子筛向多相复合分子筛的过渡阶段.

图4 m8、m16、MCM-41的SEM照片以及β分子筛的TEM照片Fig.4 SEM images of m8,m16,MCM-41 and TEM image of β zeolite(a)m8;(b)m16;(c)MCM-41;(d)β
3.3 SEM和HRTEM表征
图4为m8、m16、MCM-41的SEM照片以及β分子筛的TEM照片.纯相态复合分子筛m8的SEM图如图4(a)所示,样品表面疏松,颗粒大小均一,粒径在0.08 μm左右;图4(b)为混合相态复合分子筛m16样品,其中较大颗粒粒径在1.00 μm左右,较小颗粒的粒径在0.24 μm左右;图4(c)为MCM-41样品,呈层状结构,粒径在1.10 μm左右;图4(d)为β分子筛的TEM图,样品颗粒大小均一,粒径在0.68 μm左右.
我们以m8为纯相态样品βM的代表,以m12和m16为混相样品的代表,图5是这两种复合分子筛样品的HRTEM图,其中图5(a)为m8的HRTEM图;图5(b,c)为m12的HRTEM图;图5(d)为m16的HRTEM图.右下角插图为对应晶面的电子衍射光斑.
从图5(a)可以看出,m8有着十分规整、长程有序的六方晶体结构,其电子衍射图也很好地验证了晶体的六方结构.测量图中黑白相间的条纹的间距,得出样品的介孔孔道间距约为4.57 nm,这和XRD表征结果十分一致.该样品在(110)方向的孔道呈弧状,而单纯的MCM-41在(110)方向的孔道是直线型的,吉林大学的肖丰收认为复合分子筛特殊的弧形孔道与其特殊的制备过程有关.12在用高分辨电子透射电镜观察m8的形貌过程中,没有发现明显的微孔形貌的存在,这和XRD以及FT-IR表征结果相符.

图5 m8、m12和m16样品的HRTEM照片Fig.5 HRTEM images of m8,m12,and m16 samples(a)m8;(b,c)m12;(d)m16.The figures(b)and(c)are the HRTEM images of m12 in different scales. Insets are their corresponding beam electron diffractions.
图5(b,c)均为样品m12的HRTEM图,在观测中发现了两种不同的形貌.图5(b)和图5(a)相似,从中可以看到样品长程有序的介孔六方晶体结构和弧形的孔道走向,孔道间距为4.59 nm;而图5(c)则表现出β分子筛的微孔四方晶体结构,孔道间距为1.05 nm.除了这两种形貌外未观察到其他形貌的存在.这进一步说明m12中可能存在两种物相,分别是βM相和β相.
在m16的观测中发现m16有两种形貌,一种是β形貌,同图5(c),另外一种形貌如图5(d)所示.从图5(d)可以看到非常规整、长程有序的六方晶体结构以及非常清晰的电子衍射图,并且在(110)方向的孔道是直线型的,我们认为这不同于常规的βM物相形貌,而是更接近于MCM-41的物相形貌,造成这种形貌的可能原因是由于β晶种晶化时间过长,β晶粒尺寸较大,几乎没有参与介孔组装,只有少量β五元环结构单元参与了介孔组装,最终样品的介孔孔壁构成更接近MCM-41,所以(110)方向呈直线型走向.
从这三种样品的HRTEM图分析中,可以得到以下结论:在晶种晶化时间较短时,样品为纯相的微介孔复合分子筛βM;随着晶种晶化时间延长,样品中逐渐出现β物相,成为β物相和βM的混相;当晶种晶化时间达到一定的限值后,样品中βM相微介孔分子筛孔壁中β次级结构单元逐渐减少,并逐渐接近MCM-41物相.这个结论很好地验证了2.2节中关于复合分子筛物相的推测.
3.4 N2低温吸附等温线
图6为MCM-41、m8和m16的低温N2吸附等温线,其中图6A为p/p0=10-7-1范围内各样品的N2吸附等温线,图6B为p/p0=10-7-10-2范围内各样品的N2吸附等温线,图6C为三种样品的BJH孔径分布,图6D为样品m16的SF微孔孔径分布.从图6A可以看出,三种样品的吸附等温线走向均符合IV型吸附等温线,MCM-41和m8的滞后环更加明显,说明这两个样品的介孔结构更加显著.从图6B可以看出, m16的吸附等温线在p/p0=10-7-10-3范围内垂直上升,这与m16中存在微孔β结构有关.图6C中,三种样品的介孔孔径分布十分集中,均在2-4 nm之间.从图6D的m16微孔孔径分布可看出,m16微孔孔径分布集中,平均孔径约为0.55 nm,这和β分子筛的微孔平均孔径大小22一致.

图6 MCM-41、m8和m16样品的N2吸附等温线(A、B)和孔分布曲线(C、D)Fig.6 Nitrogen isotherms(A,B)and the pore size distributions(C,D)of MCM-41,m8,and m16 samples
MCM-41、m8和m16的孔道参数(t-plot法)列于表1.从表中可以看出,当β晶种晶化时间较短时,用β晶种组装介孔孔壁并没有为复合分子筛m8引入微孔孔道结构,只有晶种反应时间较长的m16样品出现了大量的微孔β孔道结构,其微孔面积占总比表面积的0.39.这说明,β晶种组装过程并不是β完整晶体组装成介孔孔壁,而是β的次级结构单元——五元环结构组装成为介孔孔壁,因而β晶种组装后的复合分子筛并没有引入微孔分子筛β的孔道结构,而是单纯的类似MCM-41的介孔孔道结构.
3.5 甲苯吸附性能
图7列出了相同投料硅铝比(Si/Al比为30)的β、MCM-41和复合分子筛样品对甲苯的动态吸附容量,对比了550°C焙烧样品以及在800°C下经混合气(50%水蒸气与50%空气)处理5 h所得样品的甲苯吸附性能.所有吸附数据都是在20°C的环境温度下测定的.

表1 MCM-41、m8和m16样品的孔道参数Table 1 Pore parameters of MCM-41,m8,and m16 samples
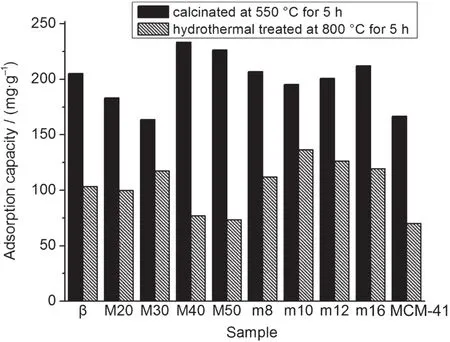
图7 β、MCM-41和β/M分子筛的甲苯吸附性能Fig.7 Toluene adsorption capacity of β,MCM-41,and β/M zeolites
稀溶液法合成的复合分子筛中,M40和M50原样对甲苯吸附容量非常高,分别是233.5和226.5 mg·g-1,超过了β的205.1 mg·g-1和MCM-41的166.6 mg·g-1;但M40和M50水热老化后对甲苯的吸附容量下降,低于同样条件下处理的β分子筛,略高于MCM-41.550°C焙烧的样品M20和M30的吸附容量和MCM-41持平;与此相反的是,M20和M30水热老化后的样品对甲苯的吸附容量与β持平或偏高,远高于MCM-41.M40和M50原样的吸附容量高的原因是二者为混相的复合分子筛,微孔孔道和介孔孔道均贡献了一定的吸附容量,但由于其晶种晶化时间较长,晶种难以参与介孔组装,因而介孔孔壁的β五元环结构较少,孔壁的热稳定性较差,在高温水热条件下孔壁坍塌,降低了其对甲苯的吸附容量;而M20和M30在晶种组装后孔容和比表面积没有显著提高,和MCM-41接近,所以其原样对甲苯的吸附容量偏低和MCM-41接近,但由于其孔壁部分晶化,由β次级结构单元组装,其水热稳定性较高,因而在水热处理后仍能保持较高的甲苯吸附容量.
浓溶液法合成的复合分子筛样品的原样对甲苯的吸附容量均比MCM-41高出许多,且略高于β;水热老化过的复合分子筛样品对甲苯吸附容量均比β和MCM-41高出许多,其中以m10的吸附容量最高,为136.4 mg·g-1.浓溶液法合成的复合分子筛的吸附性能和水热稳定性整体上比稀溶液法高,这可能是由于在晶种晶化阶段的结晶动力学决定的.较高浓度的硅铝溶胶容易形成大量粒径均一、分布均匀的β分子筛晶核,由于晶核浓度较高,晶核不易继续生长成为β晶体,而易于组装为介孔结构;在稀溶液条件下,β晶核容易继续生长成为β晶体,难以参与介孔组装.而组装后的复合分子筛样品由于拥有较大的介孔孔体积和比表面积,并保留了微孔β的酸性,提高了水热稳定性,其对甲苯的吸附性能优于单纯的微孔β和单纯的介孔MCM-41.考虑到HC-trap材料高温高湿的使用环境,5我们认为浓溶液法制备的复合分子筛样品对甲苯的吸附性能比稀溶液法更适用于汽车冷启动条件下对芳香烃的吸附.
4 结论
采用稀溶液和浓溶液两步水热合成法,通过控制第一步晶种晶化时间,制备出一系列β和MCM-41复合分子筛样品β/M,采用XRD、FT-IR和TEM测试手段表征了样品的物相结构与微观形貌,并对自组装过程进行了分析.所得结论是:复合分子筛β/M的合成过程是微孔β结构和微介孔βM结构的协同竞争反应,β晶种颗粒度大小对组装过程起重要作用,第一步晶化时间较短形成β晶种颗粒度小,最终组装形成了纯相态的微介孔复合型分子筛βM;晶化时间长形成的β晶种颗粒度大,最终形成βM和β分子筛的混合相.
选取甲苯作为探针分子分别研究了MCM-41、β以及复合分子筛对芳香烃分子的吸附性能,比较了三种分子筛及其老化后样品的甲苯吸附性能的优劣,并优选出吸附性能最好的复合分子筛m10,其老化处理后的甲苯吸附容量为136.4 mg·g-1,远高于相同条件下的MCM-41和β分子筛,有着广阔的研究和应用前景.
(1) Park,J.H.;Park,S.J.;Nam,I.S.;Yeo,G.K.;Kil,J.K.;Youn, Y.K.Microporous Mesoporous Mat.2007,101,263.
(2) Park,J.H.;Park,S.J.;Ahn,H.A.;Nam,I.S.;Yeo,G.K.;Kil,J. K.;Youn,Y.K.Microporous Mesoporous Mat.2009,117,178.
(3) López,J.M.;Navarro,M.V.;García,T.;Murillo,R.;Mastral, A.M.;Varela-Gandía,F.J.;Lozano-Castelló,D.;Bueno-López, A.;Cazorla-Amorós,D.Microporous Mesoporous Mat.2010, 130,239.
(4) Czaplewski,K.F.;Reitz,T.L.;Kim,Y.H.;Snurret,R.Q. Microporous Mesoporous Mat.2002,56,55.
(5)Burke,N.R.;Trimma,D.L.;Howe,R.F.Applied Catalysis B 2003,46,97.
(6) Liu,Y.;Pinnavaia,T.J.Studies in Surface Science and Catalysis 2002,142,1075.
(7) Triantafyllidis,K.S.;Lappas,A.A.;Vasalos,I.A.;Liu,Y.; Pinnavaia,T.J.Studies in Surface Science and Catalysis 2004, 154,2853.
(8) Triantafyllidis,K.S.;Iliopoulou,E.F.;Antonakou,E.V.; Lappas,A.A.;Wang,H.;Pinnavaia,T.J.Microporous Mesoporous Mat.2007,99,132.
(9) Triantafyllidis,K.S.;Lappas,A.A.;Vasalos,I.A.;Liu,Y.; Pinnavaia,T.J.Catalysis Today 2006,112,33.
(10) Liu,Y.;Zhang,W.;Pinnavaia,T.J.Angew.Chem.Int.Edit. 2001,40,1255.
(11) Di,Y.;Yu,Y.;Sun,Y.Y.;Yang,X.Y.;Lin,S.;Zhang,M.Y.;Li, S.G.;Xiao,F.S.Microporous Mesoporous Mat.2003,62,221.
(12) Zhang,Z.T.;Han,Y.;Xiao,F.S.;Qiu,S.L.;Zhu,L.;Wang,R. W.;Yu,Y.;Zhang,Z.;Zou,B.S.;Wang,Y.Q.;Sun,H.P.;Zhao, D.Y.;Wei,Y.J.Am.Chem.Soc.2001,123,5014.
(13) Xiao,F.S.;Han,Y.;Yu,Y.;Zhang,Z.T.;Qiu,S.L.Preparation Methods of Mesoporous Materials with StrongAcidity and Extraordinary Hydrothermal Stability.CN Patent 1349929A, 2002-05-22.[肖丰收,韩 宇,于 沂,张宗涛,裘式纶.强酸性和高水热稳定性的介孔分子筛材料及其制备方法:中国, CN1349929A[P].2002-05-22.]
(14) Van Oers,C.J.;Stevens,W.J.J.;Bruijn,E.;Mertens,M.; Lebedev,O.A.;Van Tendeloo,G.;Meynen,V.;Cool,P. Microporous Mesoporous Mat.2009,120,29.
(15) Zheng,J.L.;Zhai,S.R.;Wu,D.;Sun,Y.H.Journal of Solid State Chemistry 2005,178,1630.
(16)Zheng,J.L.;Zhang,Y.;Wei,W.;Wu,D.;Sun,Y.H.;Deng,F.; Luo,Q.;Yue,Y.Acta Phys.-Chim.Sin.2003,19,907.[郑均林,张 晔,魏 伟,吴 东,孙予罕,邓 风,罗 晴,岳 勇.物理化学学报,2003,19,907.]
(17) Ji,D.K.;Li,S.Y.;Ding,F.C.;Chi,Y.L.;Yi,Y.F.;Zhao,R.S. Journal of China University of Petroleum(Natural Science Edition)2010,34,140. [冀德坤,李术元,丁福臣,迟姚玲,易玉峰,赵如松.中国石油大学学报(自然科学版),2010,34, 140.]
(18) Bordoloi,A.;Devassy,B.M.;Niphadkar,P.S.;Joshi,P.N.; Halligudi,S.B.Journal of Molecular Catalysis A:Chemical 2006,253,239.
(19) Ma,Y.H.;Zhao,H.L.;Tang,S.J.;Hu,J.;Liu,H.L.Acta Phys.-Chim.Sin.2011,27,689. [马燕辉,赵会玲,唐圣杰,胡 军,刘洪来.物理化学学报,2011,27,689.]
(20) Li,G.;Gan,Q.B.;Zhang,H.J.;Wu,T.H.Petrochemical Technology 2002,31,106.[李 工,阚秋斌,章慧杰,吴通好.石油化工,2002,31,106.]
(21) Shih,P.C.;Wang,J.H.;Mou,C.Y.Catalysis Today 2004, 93-95,365.
(22) Prokešová,P.;Mintova,S.;Čejka,J.;Bein,T.Microporous Mesoporous Mat.2003,64,165.
(23) Perez-Pariente,J.;Martens,J.A.;Jacobs,P.A.Applied Catalysis 1987,31,35.
(24) Lin,S.Acidity and Improving Catalytic Properties in Benzene Alkylation over PreformedAluminosilicate.Ph.D.Dissertation, Jilin University,Changchun,2004. [林 森.沸石分子筛前驱体的酸性表征及催化性质研究[D].长春:吉林大学,2004.]
(25) Mintova,S.;Valtchev,V.;Onfroy,T.;Marichal,C.;Knözinger, H.;Bein,T.Microporous Mesoporous Mat.2006,90,237.
December 31,2011;Revised:March 8,2012;Published on Web:March 14,2012.
Synthesis of Combined Micro-Mesoporous Zeolites and Their Application to Automobile Cold Start Emission Control
ZHANG Lan ZHANG Zhen-Zhong WEI Ji-Ying*MENG Bi-Fang JIANG Feng LIANG Tong-Xiang
(Institute of Nuclear and New Energy Technology,Tsinghua University,Beijing Key Laboratory of Fine Ceramics, Beijing 100084,P.R.China)
Synthesis of micro-mesoporous zeolites β/M(where β and M denote β zeolite and MCM-41 zeolite,respectively)combined with zeolites β and MCM-41 was achieved in the sodium hydroxide system using a two-step hydrothermal treatment method.Sodium aluminate and fume silica were used as the sourcesofaluminum and silicon,while tetraethylammonium hydroxide (TEAOH)and cetyltrimethylammonium bromide(CTAB)were used as templates for the formation of β crystal seeds and for self-assembling of β crystal seeds into mesoporous zeolites β/M,respectively.The samples were characterized by X-ray diffraction(XRD),Fourier transform infrared(FT-IR)spectroscopy,and high resolution transmission electron microscope(HRTEM).Reaction time and particle size of β crystal seeds were found to play an important role in the synthesis of β/M zeolites.When β seeds crystallize in a short time,a pure phase(denoted βM)can result,whose mesoporous structure resembles MCM-41,while long crystallization times can result in a phase mixture of mesoporous βM and microporous β zeolite.Due to the insertion of the secondary building units of β zeolite into the mesoporous wall,the β/M micro-mesoporous combined zeolites showed enhanced toluene adsorption performance and hydrothermal stability.
Micro-mesoporous combined zeolite;Crystallization time;Grain size;Toluene adsorption;Automobile cold start emission control
10.3866/PKU.WHXB201203142
∗Corresponding author.Email:weijiying@tsinghua.edu.cn;Tel:+86-10-89796148.
The project was supported by the National Natural Science Foundation of China(21077064).
国家自然科学基金(21077064)资助项目
O647.33
