铂纳米颗粒增强MnO2纳米棒对CO和挥发性有机化合物的氧化活性
2012-12-05吴小琴宗瑞隆朱永法
吴小琴 宗瑞隆 朱永法,*
(1清华大学化学系,北京100084;2南昌航空大学环境与化学工程学院,南昌330063)
铂纳米颗粒增强MnO2纳米棒对CO和挥发性有机化合物的氧化活性
吴小琴1,2,*宗瑞隆1朱永法1,*
(1清华大学化学系,北京100084;2南昌航空大学环境与化学工程学院,南昌330063)
通过水热法合成了纯相的α-MnO2和δ-MnO2纳米棒,并利用溶胶固定化工艺制备了负载铂纳米颗粒的Pt/MnO2材料.通过透射电镜(TEM),X射线粉末衍射(XRD),扫描电镜(SEM),X射线光电子能谱(XPS),N2吸附-脱附和H2程序升温还原(H2-TPR)技术研究了样品的微观结构和吸附活性位,探查了CO和挥发性有机化合物(VOCs)(苯和甲苯)在催化剂上的催化发光(CTL)性质.结果表明:铂颗粒在α-MnO2和δ-MnO2载体上以高分散状态存在,负载过程不会影响α-MnO2纳米棒的晶相结构,但会导致δ-MnO2纳米棒产生结构变化.经XPS证实不是Pt与其发生了反应.α-和δ-MnO2纳米棒对CO、苯和甲苯的催化氧化都具有很高的活性,δ-MnO2的活性略高于α-MnO2相.虽然N2吸附-脱附实验结果证实Pt负载会导致MnO2纳米棒比表面积的下降,但H2-TPR结果显示Pt和MnO2之间会产生强烈的相互作用,显著增强其催化活性,且Pt/δ-MnO2活性高于Pt/α-MnO2.催化氧化发光研究表明,这四种催化剂活性顺序是α-MnO2≤δ-MnO2<Pt/α-MnO2<Pt/δ-MnO2,与H2-TPR结果一致.铂的负载可以显著增强α-MnO2和δ-MnO2纳米棒对CO、苯和甲苯催化氧化的活性.
催化活性;MnO2纳米棒;Pt纳米颗粒;催化发光;CO;苯;甲苯
1 引言
挥发性有机物(VOCs)不仅是有毒的空气污染物(因其毒性,恶臭,致突变和致癌性),还是臭氧和光化学烟雾的前驱体.1-3已经有多种技术应用于VOCs的减排和降解净化,其中催化氧化被公认是控制VOCs排放的有效方法之一.催化氧化可以在较低温度下对VOCs实现净化,具有能量效率高,无NOx排放,以及适用于非常稀(<5000×10-6(体积分数))污染物的直接氧化净化的特点,而这种浓度的污染物一般在热燃烧中无额外的燃料是没法处理的.4-7目前,贵金属(Pt,Pd和Au)负载催化剂8-11和金属氧化物催化剂12,13普遍被用于减少VOCs的排放.一般而言,对于VOCs的氧化净化,铂系金属是高活性的催化剂.金属氧化物负载铂复合催化剂的研究,一直是人们的兴趣.在VOCs和CO的催化燃烧中所用的载体有:Al2O3,SiO2,MgO,TiO2,SnO2,ZrO2, ZnO,CeO2,WO3-ZrO2和La0.9Cu0.1MnO3,LaCoO3,石墨烯等.14-21
MnO2是一种VOCs氧化的优良催化剂,22-24对于CO的催化氧化,其活性与MnO2纳米棒的相结构有明显关系,催化活性顺序如下:α-MnO2≈δ-MnO2>γ-MnO2>β-MnO2.25本研究基于纳米Pt对VOCs及α-和δ-MnO2纳米棒催化剂对于CO的催化氧化高活性,首次将Pt纳米颗粒与α-和δ-MnO2纳米棒组成复合催化剂,通过透射电镜(TEM)、X射线粉末衍射(XRD)、扫描电镜(SEM)、X射线光电子能谱(XPS)、N2吸附-脱附和H2程序升温还原(H2-TPR)技术(氢气消耗首次采用热导检测器(TCD)和质谱(MS)联合检测)研究催化剂结构.基于催化剂的催化发光(CTL)强度与催化活性有很好的相关性,CTL能作为快速和有效的方法扫描催化剂的活性.26直接采用CTL测定四种催化剂(α-MnO2,δ-MnO2,Pt/α-MnO2,Pt/ δ-MnO2)催化氧化CO和VOCs(以苯和甲苯作为代表VOCs的模型气体)的活性.以期获得催化剂结构与催化性能之间的关系.探索负载Pt对催化剂性能产生的影响及其根本原因.
2 实验
2.1 MnO2纳米棒催化剂的合成
α-MnO2纳米棒典型的合成过程是:将0.50 g KMnO4(北京化工厂,分析纯)和0.21 g MnSO4· H2O(北京益利精细化学品有限公司,分析纯)在蒸馏水(32 mL)中混合,磁力搅拌约10 min以形成均匀混合物.然后把混合物转移至水热釜(40 mL)中,加热140°C维持12 h.27冷却至室温,收集产品,洗涤并在80°C干燥.类似地,δ-MnO2纳米棒通过0.6 g KMnO4和0.11 g MnSO4·H2O化学氧化而得,加热至240°C维持24 h.25
2.2 铂纳米微粒/MnO2纳米棒负载催化剂的合成
胶体铂通过NaBH4(北京化工厂,分析纯)作还原剂氧化K2PtCl6(北京化工厂,分析纯)而得,并用聚乙烯醇(PVA,北京益利精细化学品有限公司,实验试剂,平均聚合度1750±50)作保护剂以达到较好的分散性.17具体过程如下:取8 mL的K2PtCl6水溶液(含Pt量为1.60 mg·mL-1),在激烈搅拌下,在冰水浴中与保护剂PVA混合(m(Pt):m(PVA)=1.5:1),然后快速注入NaBH4水溶液(n(Pt):n(NaBH4)=1:5),当溶胶从淡黄色变为深棕色,表明铂溶胶已经形成.在剧烈搅拌下,把载体MnO2纳米棒加入到铂溶胶悬浮液中,保持两者接触直至全部吸收.铂在载体中的负载量(w)为0.4%,0.8%,1.2%,1.6%.然后把湿的固体催化剂置于红外灯下烘烤直至干燥.
用于催化发光实验的陶瓷棒的制备:取催化剂200 mg和无水乙醇(北京化工厂,分析纯)完全混合至糊状后,把催化剂均匀铺在陶瓷棒表面,最后磁棒在红外灯下烘烤至干.
2.3 催化剂的表征
XRD在德国Bruker D8 Advance X射线衍射仪上进行表征,采用石墨单色器Cu靶Kα辐射(λ=0.154 nm),步宽0.02°,管电压40 kV,管电流40 mA.粉体被装进一个数据收集玻璃样品架中,扫描范围(2θ)从10°到70°,扫描速率为10(°)·min-1.
N2吸附-脱附曲线测定在美国Micromeritics公司的Tristar II 3020M化学吸附仪上进行.在-196°C (77 K)温度下,用液氮吸附法测定.测量前,样品在氮气流中300°C持续加热1.5 h以除去水分,然后在300°C持续4 h真空脱气.用BET(Brunauer-Emmett-Teller)法计算比表面积;用BJH(Barrett-Joyner-Halenda)法测定吸附总孔隙容积.
催化剂颗粒形貌和颗粒度采用TEM(JEM-1200EX,日本电子公司)观察.电子束加速电压为100 kV,样品粉末在乙醇中超声分散,然后滴在由铜网支撑的无定形碳膜上.样品的形貌和组成还通过SEM(KYKY 2800)比较.加速电压为15 kV,电流为1.2 nA.
H2-TPR实验在美国Chemisorb 2720脉冲化学吸附系统和Micromeritics TPX系统上进行.催化剂粉末(40 mg)置于“U”型石英反应器中,在120°C下用流速为50 mL·min-1氮气流冲洗1 h以除去水分(在预处理包中完成).冷却至室温后,样品用10.1% H2/Ar混合气(北京市华元气体化工有限公司)还原,流速为20 mL·min-1,升温速率为10°C·min-1直至600°C,氢气消耗同时用系统配置的TCD和串联的质谱检测器(英国Hiden QIC-20)监测.
X射线光电子能谱分析:使用ULVAC-PHI公司的PHI Quantera型X射线光电能谱仪,激发源为经单色化处理后的AlKα射线,能量为1486.7 eV.样品以污染碳的C 1s结合能(284.8 eV)作为校正基准.
2.4 催化剂的评价
催化剂的催化氧化活性用CTL作半定量评价. CTL强度用BPCL微弱发光测量仪(中国科学院生物物理研究所制造)直接测定.CTL系统原理图见参考文献.26将陶瓷催化棒放入内径为12 mm的石英管中,通过调节加热电压可以方便地控制加热棒的反应温度.从反应室入口空气以一定的流速经进样阀流过反应室,同时利用进样系统将一定量的检测气体注入进样阀,经流速为160 mL·min-1空气载入反应室,以不同波长的窄带滤光片进行不同波长信号的检测,并记录发光强度与时间的关系.
3 结果与讨论
3.1 催化剂的形貌结构
图1是合成MnO2催化剂的典型TEM图.TEM结果表明所制备的α-MnO2和δ-MnO2样品具有纳米棒的形态.α-MnO2纳米棒的直径和长度分别在13-28 nm和0.2-0.6 μm区间;δ-MnO2纳米棒的直径大约是11 nm,长度约为0.5 μm.α-,δ-MnO2催化剂的长径比分别约为11-26和50.
Pt/α-MnO2样品的TEM形貌与其前驱体相似,但Pt/δ-MnO2的TEM形貌与其前驱体有明显差别,虽仍然存在棒状结构.在这些催化剂上,Pt纳米颗粒近似球形,而MnO2主要保持其棒状.分散在α-MnO2纳米棒和δ-MnO2纳米棒上的Pt平均粒度约3-5 nm,且无明显团聚.
图2是催化剂的XRD图谱.结果表明水热合成可以获得纯相的α-和δ-MnO2结构(Fig.2(a)).负载Pt后,Pt/α-MnO2样品的XRD图与前驱体是相似的,表明Pt以高分散态负载在α-MnO2载体上.对于Pt/ δ-MnO2样品,其XRD图与其前驱体明显不同:除了仍有δ-MnO2结构外,还有新的物质Mn3O4(JCPDS 24-0734)的物相结构存在,说明部分δ-MnO2被还原.根据资料,28从10°至70°范围,有三个Pt晶面衍射峰:40.0°(111),46.5°(200)和67.4°(220).然而,在2θ区域,XRD图中都未检测到Pt峰.意味着Pt颗粒是高度分散在α-MnO2和δ-MnO2载体表面(<5 nm),这个结果和TEM一致.在Pt含量低于1.6%(w)时,不会出现Pt峰.

图1 α-,δ-MnO2纳米棒及其负载0.8%(w)Pt的TEM图谱Fig.1 TEM images of α-,δ-MnO2nanorods and 0.8%(w)Pt loading(a)α-MnO2;(b)Pt/α-MnO2;(c)δ-MnO2;(d)Pt/δ-MnO2
为了进一步证实负载Pt后δ-MnO2纳米棒结构的变化,对δ-MnO2和Pt/δ-MnO2催化剂进行了SEM测定,结果见图3.
由图3可知,δ-MnO2基本为棒(线)状.Pt/δ-MnO2催化剂既有棒状结构,也有许多其它的非一维结构.说明铂溶胶与δ-MnO2之间进行了氧化还原反应.为什么负载不会使α-MnO2发生变化(载铂量为0-1.6%(w),见Fig.2(b)),而使δ-MnO2产生改变?因为δ-MnO2与其它一维MnO2纳米结构不同,不是隧道结构而是层状结构.它的管状结构是在高温下形成的,在负载过程的低温处理中可能会展开重新形成层状结构.层状结构是一个亚稳定状态,如果有足够的K+支撑,δ-MnO2将保持其结构;27可能是NaBH4中Na+的引入取代了部分K+,使部分层状结构崩溃并与体系存在的还原性物质发生氧化还原反应,使δ-MnO2得到电子形成Mn3O4.XRD结果与TEM和SEM结果是一致的.
3.2 催化剂的比表面积,孔容积和晶粒度影响
四种催化剂的比表面积,孔容积和晶粒度数据见表1.通过Scherrer公式计算,α-,δ-MnO2和Pt/α-, δ-MnO2纳米棒的平均晶粒度依次为20.5、20.5、11和11 nm.此外,α-MnO2、Pt/α-MnO2、δ-MnO2和Pt/δ-MnO2纳米棒催化剂的BET比表面积分别为47、28、94和63 m2·g-1,δ-MnO2纳米棒的BET比表面积比α-MnO2纳米棒更大,当Pt负载后,α-MnO2和δ-MnO2的比表面积和孔容积都有明显降低.
比表面积通常是影响催化剂活性的主要因素,一般比表面积与催化剂的活性具有正相关规律.在此,δ-MnO2纳米棒催化剂的BET比表面积(94 m2· g-1)比α-MnO2(47 m2·g-1)大一倍,前者的催化活性略高于后者.虽然负载铂后比表面积都下降了,但Pt/ δ-MnO2BET比表面积(63 m2·g-1)还是比Pt/α-MnO2的BET比表面积(28 m2·g-1)大,所以前者的催化活性还是大于后者.但不同类型(有或无负载Pt)的催化剂相比较,小比表面积负载铂催化剂活性比未负载的催化剂大.这说明比表面积并不是决定催化活性的主要因素.本结论与文献25的结果一致.
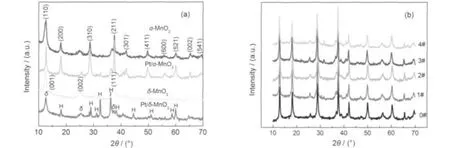
图2 α-,δ-MnO2纳米棒及其负载0.8%Pt(a)及不同负载量的Pt/α-MnO2(b)的XRD图谱Fig.2 XRD patterns of α-,δ-MnO2nanorods and 0.8%Pt loading(a)and Pt/α-MnO2with different Pt loadings(b)0#,1#,2#,3#,4#in Fig.2(b)represent 0,0.4%,0.8%,1.2%,1.6%(w)Pt in the Pt/α-MnO2nanorods,respectively.

图3 δ-MnO2纳米棒(a)和Pt/δ-MnO2催化剂(b)的SEM图Fig.3 SEM images of δ-MnO2nanorods(a)and Pt/δ-MnO2catalysts(b)

表1 MnO2纳米棒的比表面积、孔体积、晶粒度及长径比Table 1 Specific surface area,porous volume,crystal size,and aspect ratio
3.3 MnO2和Pt/MnO2的H2-TPR分析
α-MnO2,Pt/α-MnO2,δ-MnO2和Pt/δ-MnO2纳米棒的H2-TPR测定结果如图4所示.由图4可见,α-MnO2纳米棒有两个H2消耗峰,292和322°C(曲线a);δ-MnO2纳米棒也有两个还原峰,263和316°C (曲线c),与α-MnO2相似,但向低温区移动.这个结果表明δ-MnO2比α-MnO2纳米棒更易还原,具有更低温度的催化氧化活性位.低温峰应该归因于MnO2还原为Mn3O4,高温峰应该归因于Mn3O4还原至MnO.29MnO2纳米棒的主要还原产物是MnO,这可以通过在H2-TPR测试后,棕红色α-MnO2和黑色δ-MnO2都变为绿色而证实.然而,Pt/α-MnO2和Pt/ δ-MnO2TPR图与α-MnO2和δ-MnO2的有很大的不同.与α-MnO2比较,Pt/α-MnO2的TPR峰移至更低温度,并且能观察到三个H2消耗峰,135、211和377°C (曲线b).135°C的峰应该归因于与Pt密切接触的表面的锰的还原,211°C的高温峰,归因于未与Pt接触的表面锰的还原,而377°C峰是由体相锰的还原产生.7当Pt负载在δ-MnO2纳米棒后,也能观察到三个还原峰在123、249和348°C(曲线d),但峰形与Pt/α-MnO2的TPR不同,第一个峰面积更小,表示与铂接触的δ-MnO2比α-MnO2更少,高温区的峰面积更大,表示Mn3O4还原至MnO的量更多.这间接证明了Pt/δ-MnO2催化剂本身就存在Mn3O4.总之,Pt和MnO2间有强烈的相互作用发生,通过从Pt原子到氧化锰的氢溢流促进MnO2还原.30这个结果表明四种催化剂还原增强顺序为:α-MnO2<δ-MnO2<Pt/ α-MnO2<Pt/δ-MnO2.在H2-TPR测定实验中,MS监测结果与TCD结果一致(图4(b)).
3.4 Pt/MnO2催化剂表面离子价态

图4 α-,δ-MnO2纳米棒及其负载0.8%Pt的H2-TPR图Fig.4 H2-TPR profiles of α-,δ-MnO2nanorods and 0.8%Pt loading(A)monitored by TCD,(B)monitored by MS;(a)α-MnO2,(b)Pt/α-MnO2,(c)δ-MnO2,(d)Pt/δ-MnO2

图5 Pt/MnO2样品中O 1s(A),Mn 2p(B),Pt 4f(C)的XPS图谱Fig.5 O 1s(A),Mn 2p(B),and Pt 4f(C)XPS spectra of Pt/MnO2(a)Pt/α-MnO2,(b)Pt/δ-MnO2
为探讨Pt/δ-MnO2催化剂中部分δ-MnO2还原为Mn3O4的原因,对负载样品进行了XPS测定,图5为样品Pt/α-MnO2和Pt/δ-MnO2的O 1s、Mn 2p和Pt 4f的XPS谱图.图5(A)显示在Eb≈530.0 eV处存在一个O 1s峰,它对应着氧物种Oα,为表面晶格氧物种(O2-);在Eb≈531-532.5 eV处也有一个O 1s宽峰,它对应着氧物种Oβ,为表面吸附氧物种(O或O-),24其主要来源于Pt/MnO2样品表面氧空位对氧的吸附. 531 eV处为OH-,而532.5 eV处则为H2O.31表明Pt/ MnO2与Au/MnO2类似,也存在着金属-载体的相互作用并导致表面羟基富集和吸附水积累.23,32从图5 (A)还可看出:Pt/δ-MnO2比Pt/α-MnO2有更多的吸附氧物种.
图5(B)显示两种样品的Mn 2p3/2峰,结合能约为642.3 eV,它与文献33报道MnO2的Eb=642.0±0.2 eV基本吻合.Mn 2p1/2峰位于Eb≈654.1 eV.Mn 2p的自旋轨道分裂能约为11.8 eV,这与MnO2和Mn2O3相似.34即样品中都存在Mn3+和Mn4+物种.645和646 eV肩峰特征是由于低价锰物种的存在,即MnO或Mn3O4.31,35在此Pt/δ-MnO2上未见与Pt/α-MnO2的明显不同.而在前面的XRD图(图2(a))中它们两者是略有差别的.这是因为XRD涉及晶体结构,XPS只与价态有关.
图5(C)显示Pt的4f7/2(Eb=71.4 eV)和4f5/2(Eb= 74.8 eV)两个峰,说明Pt/δ-MnO2与Pt/α-MnO2两种样品表面只有零价态的金属铂存在.与La0.9Cu0.1MnO3和LaCoO3载体一样.16铂与载体间没有发生氧化还原反应.
为探究Pt/δ-MnO2催化剂中部分δ-MnO2还原为Mn3O4的原因,做了如下实验:用合成Pt/δ-MnO2催化剂同样的方法(不加K2PtCl6)处理δ-MnO2,查看是否是NaBH4使部分δ-MnO2还原,XRD结果证实不是.在此推测是氢气的作用,因为负载过程存在如下反应:

负载过程也发现,在α-MnO2样品中白色气泡很快消失,而δ-MnO2样品中白色气泡消失较慢.
3.5 催化氧化发光性质研究
3.5.1 铂负载量对CTL强度的影响
因为CTL强度与催化活性有较好的相关性,因此CTL能够作为催化剂活性的快速测量手段.利用负载不同量(0-1.6%)Pt颗粒的α-MnO2复合催化剂,对CO、苯和甲苯的催化氧化发光性能进行了研究,结果见图6.
以上CTL结果表明,Pt/α-MnO2催化剂随Pt负载量变化催化氧化CO、苯和甲苯的活性规律相似,其活性顺序如下:Pt/α-MnO2(0.8%)>Pt/α-MnO2(0.4%)>Pt/α-MnO2(1.2%)>Pt/α-MnO2(1.6%)>α-MnO2.且最大发射波长均为640 nm,即所有反应物均被催化氧化为同样的产物——CO2.

图6 不同Pt负载量对CTL强度和最大发射波长的影响Fig.6 Effect of different Pt loadings on CTLintensity and biggest CTLemission wavelengthv=160 mL·min-1,T=260°C,injection sample concentration:143 μg·mL-1;(a)CO,(b)benzene,(c)toluene

图7 在四种催化剂上催化发光强度随温度的变化Fig.7 CTLintensity as a function of temperature over the four catalystsconditions:catalyst,0.8%Pt/MnO2;v=200 mL·min-1;injection sample:20 mL saturated vapor(25°C); (a)CO,(b)benzene,(c)toluene
3.5.2 反应温度对CTL强度的影响
图7显示了CO、苯和甲苯在不同温度下,分别在α-MnO2、Pt/α-MnO2、δ-MnO2和Pt/δ-MnO2纳米棒上的CTL响应.在四种催化剂上,CTL强度随着温度升高而增加.能观察到在Pt/δ-或Pt/α-MnO2上的CTL强度比在δ-或α-MnO2纳米棒上高.在CO和苯的氧化中,在整个温度测定范围内,在δ-MnO2纳米棒上和在α-MnO2上的CTL强度明显是一致的.可是,对于甲苯的氧化,α-MnO2明显不如δ-MnO2,在整个温度测定范围内,在δ-MnO2纳米棒上CTL强度基本与Pt/α-MnO2相同.总之,在四种催化剂上CTL强度增加的顺序是:α-MnO2≤δ-MnO2<Pt/α-MnO2<Pt/ δ-MnO2,这正好与H2-TPR的结论一致.α-MnO2与δ-MnO2的CTL强度基本相当,而Pt/δ-MnO2的CTL强度明显大于Pt/α-MnO2,可能是因为Mn3O4的贡献,因为它也具有良好的CTL性能.36当CO、苯和甲苯在催化剂表面被催化氧化为CO2时,释放的能量能够被一些CO2分子所吸收.其结果是,CO2分子能够从基态跃迁至激发态,当CO2分子从激发态返回至基态时,很弱的CTL光将被发射.37CTL强度与产生的CO2浓度成正比,因此,CTL对CO、苯和甲苯的响应能够证明MnO2纳米棒的催化活性.14
图7结果进一步证实,Pt促进了α-MnO2和δ-MnO2纳米棒的还原,Pt和MnO2间有强烈的交互作用发生,Pt通过其原子到氧化锰的氢溢流促进MnO2还原.因此,Pt/MnO2催化剂改进了催化活性,这和负载金30的情况一致.
4 结论
(1)CTL结果证明负载铂后,α-MnO2和δ-MnO2纳米棒的催化活性明显增加.四种催化剂还原能力增强的顺序是:α-MnO2≤δ-MnO2<Pt/α-MnO2<Pt/δ-MnO2.
(2)TPR结果与之对应:TCD和MS谱均证实负载铂后催化剂除了保持原来二氧化锰的还原表面活性位外,还增加了两个反应活性位,与铂密切接触的表面锰的还原,本体二氧化锰的还原,证实铂与载体之间存在着明显的相互作用.
(1)Amann,M.;Lutz,M.J.Hazard.Mater.2000,78,41.
(2) Li,N.;Gaillard,F.Appl.Catal.B:Environ.2009,88,152.
(3)Aguero,F.N.;Barbero,B.P.;Gambaro,L.;Cadús,L.E.Appl. Catal.B:Environ.2009,91,108.
(4) Li,Y.;Zhang,X.;He,H.;Yu,Y.;Yuan,T.;Tian,Z.;Wang,J.; Li,Y.Appl.Catal.B:Environ.2009,89,659.
(5) Gandhe,A.R.;Rebello,J.S.;Figueiredo,J.L.;Fernandes,J.B. Appl.Catal.B:Environ.2007,72,129.
(6) Liotta,L.F.Appl.Catal.B:Environ.2010,100,403.
(7)Li,H.F.;Lu,G.Z.;Dai,Q.G.;Wang,Y.Q.;Guo,Y.;Guo,Y.L. Appl.Catal.B:Environ.2011,102,475.
(8) Diehl,F.;Barbier,J.,Jr.;Duprez,D.;Guibard,I.;Mabilon,G. Appl.Catal.B:Environ.2010,95,217.
(9) He,C.;Li,J.;Li,P.;Cheng,J.;Hao,Z.;Xu,Z.P.Appl.Catal. B:Environ.2010,96,466.
(10) Pitkäaho,S.;Ojala,S.;Maunula,T.;Savimäki,A.;Kinnunen,T.; Keiski,R.L.Appl.Catal.B:Environ.2011,102,395.
(11) Ousmane,M.;Liotta,L.F.;Carlo,G.D.;Pantaleo,G.;Venezia, A.M.;Deganello,G.;Retailleau,L.;Boreave,A.;Giroir-Fendler,A.Appl.Catal.B:Environ.2011,101,629.
(12) Kim,S.C.J.Hazard.Mater.B 2002,91,285.
(13) Rivas,B.;López-Fonseca,R.;Gutiérrez-Ortiz,M.;Giérrez-Ortiz,J.I.Appl.Catal.B:Environ.2011,101,317.
(14)Wang,X.;Na,N.;Zhang,S.C.;Wu,Y.Y.;Zhang,X.L.J.Am. Chem.Soc.2007,129,6062.
(15) Comotti,M.;Li,W.C.;Spliethoff,B.;Schüth,F.J.Am.Chem. Soc.2006,128,917.
(16) Bulgan,G.;Zong,R.L.;Liang,S.H.;Yao,W.Q.;Zhu,Y.F. Acta Phys.-Chim.Sin.2008,24,1547.[Bulgan G.,宗瑞隆,梁淑惠,姚文清,朱永法.物理化学学报,2008,24,1547.]
(17) Zhang,C.;He,H.Catal.Today 2007,126,345.
(18) Beauchet,R.;Mijoin,J.;Batonneau-Gener,I.;Magnoux,P. Appl.Catal.B:Environ.2010,100,91.
(19) Wu,X.Q.;Zong,R.L.;Mu,H.J.;Zhu,Y.F.Acta Phys.-Chim. Sin.2010,26,3002.[吴小琴,宗瑞隆,牟豪杰,朱永法.物理化学学报,2010,26,3002.]
(20) Song,Y.Q.;Kang,C.L.;Feng,Y.L.;Liu,F.;Zhou,X.L.; Wang,J.A.;Xu,L.Y.Catal.Today 2009,148,63.
(21) Mitsui,T.;Tsutsui,K.;Matsui,T.;Kikuchi,R.;Eguchi,K.Appl. Catal.B:Environ.2008,78,158.
(22) Lahousse,C.;Bernier,A.;Grange,P.;Delmon,B.; Papaefthimiou,P.;Ioannides,T.;Verykiosy,X.J.Catal.1998, 178,214.
(23) Lee,S.J.;Gavriilidis,A.;Pankhurst,Q.A.;Kyek,A.;Wagner, F.E.;Wong,P.C.L.;Yeung,K.L.J.Catal.2001,200,298.
(24) Hamoudi,S.;Larachi,F.;Adnot,A.;Sayari,A.J.Catal.1999, 185,333.
(25) Liang,S.H.;Teng,F.;Bulgan,G.;Zong,R.L.;Zhu,Y.F. J.Phys.Chem.C 2008,112,5307.
(26)Teng,F.;Yao,W.Q.;Zhu,Y.F.;Chen,M.D.;Wang,R.H.; Mho,S.;Meng,D.D.J.Phys.Chem.C 2009,113,3089.
(27) Wang,X.;Li,Y.D.Chem.Eur.J.2003,9,300.
(28) Chakraborty,S.;Raj,C.R.Sensors and Actuators B 2010,147, 222.
(29)Xu,R.;Wang,X.;Wang,D.S.;Zhou,K.B.;Li,Y.D.J.Catal. 2006,237,426.
(30)Wang,L.C.;Liu,Y.M.;Chen,M.;Cao,Y.;He,H.Y.;Fan,K. N.J.Phys.Chem.C 2008,112,6981.
(31) Banerjee,D.;Nesbitt,H.W.Geochim.Cosmochim.Acta 2001, 65,1703.
(32)Wang,L.C.;He,L.;Liu,Q.;Liu,Y.M.;Chen,M.;Cao,Y.;He, H.Y.;Fan,K.N.Appl.Catal.A:Gen.2008,344,150.
(33) Kapteijn,F.;van Langeveld,A.D.;Moulijn,J.A.;Andreini,A.; Vuurman,M.A.;Turek,A.M.;Jehng,J.M.;Wachs,I.E. J.Catal.1994,150,94.
(34) Muilenbergy,G.E.Handbook of X-Ray Photoelectron Spectroscopy;Perkin-Elmer Corporation:Minnesota,1979.
(35) Srinivasan,B.;Gardner,S.D.Surf.Interface Anal.1998,26, 1035.
(36) Zhang,L.C.;Zhou,Q.;Liu,Z.H.;Hou,X.D.;Li,Y.B.;Lv,Y. Chem.Mater.2009,21,5066.
(37) Breysse,M.;Claudel,B.;Faure,L.;Guenin,M.;Williams,R.J. J.;Wolkenstein,T.J.Catal.1976,45,137.
September 29,2011;Revised:December 1,2011;Published on Web:December 8,2011.
Enhanced MnO2Nanorods to CO and Volatile Organic Compounds Oxidative Activity by Platinum Nanoparticles
WU Xiao-Qin1,2,*ZONG Rui-Long1ZHU Yong-Fa1,*
(1Department of Chemistry,Tsinghua University,Beijing 100084,P.R.China;2College of Environmental and Chemical Engineering,Nanchang Hang Kong University,Nanchang 330063,P.R.China)
Pure-phase α-MnO2and δ-MnO2nanorods were synthesized through an easy solution-based hydrothermal method.Platinum nanoparticles supported by the obtained MnO2nanorods were prepared by the colloid deposition process.The microstructure and adsorption activity of the obtained catalysts were researched by different techniques such as transmission electron microscopy(TEM),powder X-ray diffraction(XRD),scanning electron microscopy(SEM),X-ray photoelectron spectroscopy(XPS),N2adsorption-desorption measurements,and H2temperature-programmed reduction (H2-TPR).The cataluminescence(CTL)properties of CO and volatile organic compounds(VOCs),such as benzene and toluene,on the resultant catalysts were explored.The results showed that the platinum nanoparticles were well distributed in α-MnO2and δ-MnO2.In addition,the Pt load process does not affect the crystal phase structure of the α-MnO2nanorods,but can generate structural changes in the δ-MnO2nanorods.The phase transformation did not the result of the reaction between the δ-MnO2nanorods and Pt as shown in the XPS study.The α-MnO2and δ-MnO2nanorods showed a high catalytic oxidative activity toward CO,benzene, and toluene,and δ-MnO2showed a higher activity than the α-MnO2phase.Although,the Pt load led to a decrease in the surface area of the MnO2nanorods which was confirmed by the N2adsorption-desorption measurements,but the H2-TPR results showed that the interaction between Pt and MnO2was intense, which significantly enhanced its catalytic activity.The Pt/δ-MnO2nanorods exhibited a higher activity than Pt/α-MnO2.CTL research showed that the activities of the four catalysts increased in the order of α-MnO2≤δ-MnO2<Pt/α-MnO2<Pt/δ-MnO2,and the H2-TPR results were consistent.Pt loading significantly enhanced the catalytic oxidative activity of α-MnO2and δ-MnO2nanorods to CO,benzene,and toluene.
Catalytic activity;MnO2nanorod;Pt nanoparticle;Cataluminescence;CO; Benzene;Toluene
10.3866/PKU.WHXB201112082 www.whxb.pku.edu.cn
*Corresponding authors.ZHU Yong-Fa,Email:zhuyf@tsinghua.edu.cn;Tel:+86-10-62787601.WU Xiao-Qin,Email:wxq968@sina.com; Tel:+86-791-83963377.
The project was supported by the National Natural Science Foundation of China(20925725),National Key Basic Research Program of China(973) (2007CB613303),and Jiangxi Provincial Department of Education Technology Project,China(GJJ11507).
国家自然科学基金(20925725),国家重点基础研究发展规划项目(973)(2007CB613303)和江西省教育厅科技项目(GJJ11507)资助
O643;O644
