MultiSite Gateway技术在疟原虫条件性基因打靶载体快速构建中的应用
2012-12-03王各各李娇娜杜峰邓舒梁佳元曹雅明罗恩杰
王各各,李娇娜,杜峰,邓舒,梁佳元,曹雅明,罗恩杰
(1.中国医科大学基础医学院免疫学教研室,沈阳110001;2.沈阳市中医药学校基础教研室,沈阳110001;3.中国医科大学基础医学院病原生物学教研室,沈阳110001)
疟疾作为一种严重威胁人类生命健康的感染性寄生原虫疾病,据2011年WHO报告,2010年全球约33亿人受到疟疾的威胁,导致约65.5万患者死亡[1],给全球尤其是发展中国家带来了严重的健康危害和经济负担。由于疟原虫和传播蚊虫的耐药问题[1,2]及目前尚无有效的疟疾疫苗[3],所以疟疾的防治工作形势仍然十分严峻。而随着各种疟原虫基因序列相继公布,基因修饰技术(genetic modification technologies)广泛地应用于疟原虫基因功能的研究,疟原虫生物学行为及其与宿主的关系等方面的研究,为抗疟疫苗的研制和疟原虫耐药机制研究开创了新途径。对基因操纵的过程是一个繁琐又漫长的过程,除了源于转染效率较低外,还在于构建一个富含AT序列的打靶载体也很费时[4]。而MultiSite Gateway技术(MultiSite Gateway technology)为构建含有这样序列的打靶载体提供了快速方便的方法。MultiSite Gateway克隆技术的基础是lambda噬菌体的位点特异性重组反应,是一种准确保存遗传信息的有效的自然生化途径。本方法可以使DNA片段在含有重组位点的两质粒间进行定向置换,也可以用含有重组位点的DNA片段置换相应质粒上的片段。与传统的需要DNA限制性内切酶、DNA连接酶、凝胶电泳和DNA片段纯化等多个步骤的单调乏味的亚克隆方法相比,该方法操作方便、快捷,节省了大量的时间和劳力。本文以构建约氏疟原虫红细胞结合样蛋白(erythrocytebinding-likeprotein,ebl)[5],条件性敲除基因打靶载体为例,展示该方法的实施策略、操作过程及其优越性。
1 材料与方法
1.1 材料
约氏疟原虫致死型(P.yoelii17xl)、质粒pCHD43 (Calmodulin5′driving hDHFR,referring to the orientation of the att cassette and 4/3 for attR sites 4 and3,前期构建包括药物筛选基因hDHFR和基因重复reg20序列等元件)和P.bUIS4/flp[6]由长崎大学热带医学研究所提供,E.coliJM109和DH5α由本教研室保存。

1.2 方法
1.2.1 质粒pCHD-EBL-FRT的构建
主要应用MultiSite Gateway方法,完成若干DNA片段的定性重组。MultiSite Gateway方法主要包括BP反应(BP recombination reaction)和LR反应(LR recombination reaction)。BP反应,以attB为基础(attB PCR产物或是表达克隆)与attP(供体)反应后建立1个包含attL的PENT克隆。LR反应,1个包含attL的PENT克隆与1个包含attR的目的载体反应后建立一个包括attB的表达克隆。
1.2.1.1 获取EBL基因同源臂
1.2.1.1.1 PCR获取3′同源臂
以P.y17xl株基因组为模板,用分别含有F3序列、attB4位点和 SpeⅠ酶切位点、attB1位点的yEBL3UF3B4F和yEBL3USpe B1R引物(表1)扩增3′同源臂F3-EBL3U。PCR条件为94℃2 min;98℃10 s,65 ℃30 s,68 ℃30 s,35 cys;68 ℃ 7 min。在此我们使用由FRT序列修饰而来的F3序列,代替FRT,从而防止与PyUIS4/flp质粒单交叉法插入基因组(达到条件表达flp酶的目的)后所保留的FRT位点之间发生位点特异的重组。
1.2.1.1.25′同源臂的获取及pB12F3-EBL5U.orf质粒的构建
引物 FRT-F3 PstF、FRT-F3 BamR(表1)经 92 ℃变形72℃退火得F3片段,并把此片段连接到PstI,BamHⅠ酶切的pB12-2质粒上得质粒pB12F3。以P.y17xl基因组为模板,用引物yEBL5U1SpeF和yEBL5U1R(见表1)PCR 扩增EBL.5U1(577bp)片段,条件为 94 ℃2min,98℃10 s,55 ℃30 s,68 ℃1min,35 cys;68℃7 min。用一步酶切连接法完成EBL.5U1片段和PB12F3质粒的酶切和连接反应,反应体系内同时加入StuI酶和T4连接酶,经16℃2 h,25 ℃1h,6cys;65 ℃10 min;25 ℃过夜完成反应。连接质粒(PB12F3-EBL5U1)转化DH5α细胞。用菌落PCR鉴定后再以引物M13F/M13R测序验证。应用同样步骤将PCR扩增的EBL-5U2+EBL.orf(3285 bp)序列(引物yEBL5U2F和yEBLorf R;条件94℃2 min;98 ℃10 s,52 ℃30 s,68 ℃2min,35 cys;68 ℃7 min),以SmaI酶切位点的一步酶切连接法构建质粒pB12F3-EBL5U.orf,随之转化、菌落PCR和测序鉴定。
1.2.1.2 BP反应获取含有同源臂的Entry载体
PCR产物F3-EBL3U与载体pDonrP4P1R进行BP反应构建质粒pENT41F3-EBL3U。pB12F3-EBL5U.orf与pDonr221进行BP反应构建Entry载体pENT12F3-EBL5Uorf。以上反应均按Invitrogen说明书操作步骤进行。
同时构建1个含有Myc标签的质粒pENT23-6Mycs。可使置换的ebl基因C端附有Myc标签,方便ebl表达监测。

1.2.1.3 LR反应及多基因片段的组装
将3个 Entry载 体 pENT41F3-EBL3U、pENT12F3-EBL5Uorf和pENT23-6Mycs与pCHD43进行LR反应,得到目的质粒pCHD-EBL-FRT(总体构建过程见图1)。LR反应按Invitrogen说明书进行操作。随后进行转化,筛选阳性克隆,所提质粒用SpeⅠ、PstⅠ和EcoRⅠ酶切鉴定。
1.2.2 P.yUIS4/flp质粒的构建
1.2.2.1 同源臂的获取
以P.y17xl株基因组为模板,PCR扩增uis4基因的5′非翻译端作为同源臂片段(PyUIS4-5U)。并通过AT克隆构建含有PyUIS4-5U片段的质粒PGEM-T-PyUIS4-5U。通过位点特异突变(所用引物为表中yUIS4-5U.NhAv2R和yUIS4-5U.NhAv2F),使质粒PGEM-T-PyUIS4-5U的PyUIS4-5U区段含有AvrⅡ和NheⅠ两种酶切位点作为线性化位点。
1.2.2.2 同源臂的置换
用含有AvrⅡ和NheⅠ酶切位点PyUIS4-5U片段置换质粒PbUIS4/flp同源臂(PbUIS4-5U),得约氏疟原虫插入质粒PyUIS4/flp。经SmaⅠ和NcoⅠ酶切、凝胶电泳、回收、连接等多步亚克隆完成。
2 结果
2.1 同源臂序列的测序验证
质粒 pENT41F3-EBL3U、PB12F3-EBL5U1、pB12F3-EBL5U.orf和pGEM-T-PyUIS4-5U经测序显示同源臂序列与预期结果一致。
2.2 质粒酶切鉴定
2.2.1 质粒pCHD-EBL-FRT酶切鉴定:质粒pCHDEBL-FRT经SpeⅠ酶切后产生一个11.3 kb的片段;经PstⅠ酶切后产生两个片段,分别为4.6 kb和6.7 kb;经EcoRⅠ酶切后产生3个片段,分别为2.4 kb、3.6 kb和5.2 kb,均与预期结果完全一致(图2)。

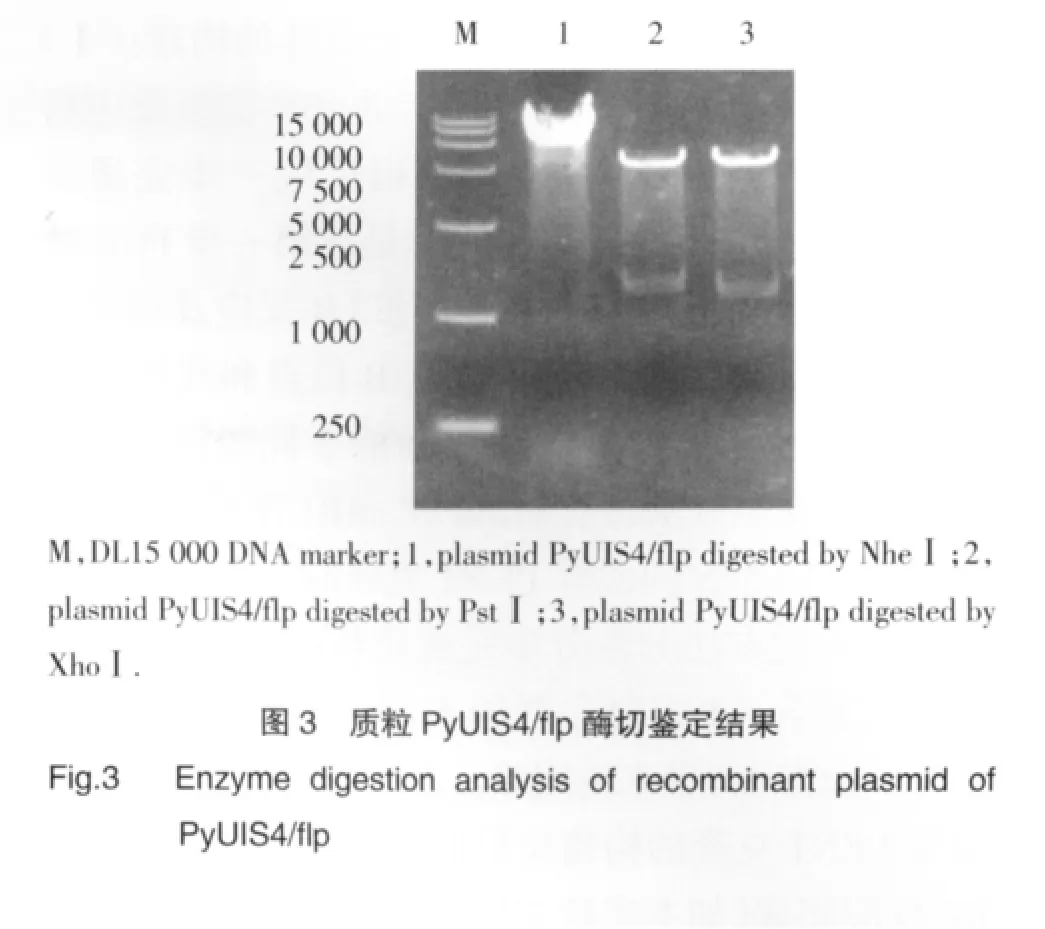
2.2.1 质粒PyUIS4/flp酶切鉴定:质粒PyUIS4/flp经NheⅠ酶切后产生一个8 kb的片段;经PstⅠ酶切后产生两个片段,分别为2.2 kb和5.7 kb;经XhoⅠ酶切后也产出两个片段,分别为1.6 kb和6.1 kb,均与预期结果完全一致(图3)。
3 讨论
随着反向基因技术(reverse genetic technologies)广泛地用于疟原虫基因功能的研究,如何快速有效地构建基因打靶载体越来越引起人们的重视。一个完整的传统型基因打靶载体一般包括:基因组同源区长、短臂,可进行原核及真核筛选的标记基因,以及打靶质粒骨架等组成部分;条件性基因敲除打靶载体除了以上的组成部分外,还需连接两侧附带loxP或FRT位点的打靶序列。目前比较常用的基因打靶载体构建方法是:先通过PCR扩增各组分,然后再分别进行酶切、连接和转化筛选。运用这种方法,即便是最普通的传统型基因敲除打靶载体也需要至少3次酶切、连接和转化反应[7]。不仅操作相对烦琐,而且多次的体外酶切与连接反应还增加了DNA突变的危险。
由于疟原虫基因组AT含量较高,使其打靶载体含有富集AT的基因片段,加之打靶载体一般较大(10 kb以上),导致打靶载体在大肠杆菌内的拷贝数较少并且固有的稳定性降低[4]。因此相对其他真核细胞打靶载体构建,疟原虫打靶载体的构建更费时费力。而MultiSite Gateway技术不需要酶切和连接反应,并且BP和LR反应比传统的连接反应有更高的效率和特异性,还可同时进行多个相互独立的位点特异的重组反应,一步即可组装4段基因片段(1段质粒骨干区和3段插入序列,如图1所示),完成质粒的构建。比传统打靶载体构建省时省力。同时考虑可用于筛选基因修饰疟原虫的耐药基因较少的情况[8,9],把耐药基因加入质粒 pCHD43 内,作为质粒骨干区与同源臂进行组装,进一步优化了质粒的构建。3步即可完成疟原虫打靶载体的构建:(Ⅰ)PCR获取相应基因的同源臂;(Ⅱ)BP重组反应得PENT克隆;(Ⅲ)通过LR重组反应一步完成多DNA片段的组装。需要注意的是在第一步PCR获取同源臂时,应考虑后续的BP和LR反应及构建打靶载体的类型设计含有相应attB位点和线性化酶切位点的引物,例如用于本实验的5′同源臂的上游和下游引物应分别含有attB4和attB1序列等。再次本实验在构建5′同源臂PENT克隆时,我们使用传统质粒构建方法分段分步完成质粒的构建,克服由较长的富含AT重复序列的5′同源臂及要加入speⅠ和F3序列所带来的困难。由此我们看出,对于同源臂PENT克隆的构建应根据实验的具体情况一步BP反应完成(如本实验3′同源臂PENT克隆载体的构建)或结合其他方法分步完成(如本实验5′同源臂PENT克隆载体的构建)。
总之,从总体操作来看,以MultiSite gateway技术为基础的体系可作为高效打靶载体的构建工具,提高基因打靶技术研究疟原虫基因功能的效率。继而我们将通过单交叉法,将PyUIS4/flp插入到UIS4基因5′非翻译区,使flp酶能在子孢子期表达,为条件敲除目的基因提供基础。再通过双交叉法,用质粒pCHD-EBL-FRT中含有F3序列的ebl基因置换原基因组ebl区段,从而达到条件敲除ebl基因的目的。最终为疟原虫与宿主的关系及减毒活疫苗的研究奠定基础。
[1]World Health Organization.World malaria report2011[M].Geneva:World Health Organization,2011:ⅹⅲ.
[2]World Health Organization.Global malaria control and elimination:Report of a meeting on containment of artemisinin tolerance[M].Geneva:World Health Organization,2008:1-3.
[3]Schwartz L,Brown GV,Genton B,et al.A review of malaria vaccine clinical projects based on the WHO rainbow table[J].Malar J,2012,11:11.
[4]Tonkin CJ,van Dooren GG,Spurck TP,et al.Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method[J].Mol Biochem Parasitol,2004,137(1):13-21.
[5]Tham WH,Healer J,Cowman AF.Erythrocyte and reticulocyte binding-like proteins of Plasmodium falciparum[J].Trends Parasitol,2012,28(1):23-30.
[6]Combe A,Giovannini D,Carvalho TG,et al.Clonal conditional mutagenesis in malaria parasites[J].Cell Host Microbe,2009,5(4):386-396.
[7]Liu HQ,Da JX,Yu WG,et al.Progress on gene targeting[J].Yi Chuan,2002,24(6):707-711.
[8]Janse CJ,Ramesar J,Waters AP.High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei[J].Nat Protoc,2006,1(1):346-356.
[9]Janse C J,Franke-Fayard B,Mair GR,et al.High efficiency transfection of Plasmodium berghei facilitates novel selection procedures[J].Mol Biochem Parasitol,2006,145(1):60-70.
