L-缬氨酸旋光异构的两种光反应可能途径
2012-11-30马驰骋李军男李志宏
马驰骋 蒲 敏,* 卫 敏 李军男 李志宏
(1北京化工大学,化工资源有效利用国家重点实验室,北京100029; 2中国科学院高能物理研究所同步辐射实验室,北京100049)
L-缬氨酸旋光异构的两种光反应可能途径
马驰骋1蒲 敏1,*卫 敏1李军男1李志宏2
(1北京化工大学,化工资源有效利用国家重点实验室,北京100029;2中国科学院高能物理研究所同步辐射实验室,北京100049)
利用密度泛函理论(DFT)和从头算分子轨道理论对L-缬氨酸的旋光异构光反应机理进行了研究.分别用B3LYP和MP2方法在6-311++G(d,p)基组级别上全优化得到了S0和T1态反应路径上的反应物、产物、中间体以及过渡态结构的几何构型,给出了反应能垒,利用含时密度泛函理论(TD-DFT)中的B3LYP/6-311++G(d, p)方法优化得到了S1态反应路径上的平衡态几何构型.通过分析反应途径上各个驻点的几何构型特征,确定了L-缬氨酸在激发态可能通过手性碳上的氢原子以羰基氧或氨基氮为中转媒介发生质子迁移来完成旋光异构反应.进一步用自洽反应场理论中的极化连续模型(PCM)方法探讨了溶剂化效应对旋光异构反应机理的影响.
缬氨酸;旋光异构;密度泛函理论;从头算分子轨道理论;溶剂化效应
1 引言
氨基酸消旋现象的研究在化学及生命科学中是一个十分重要且复杂的课题,对确定化石年代、预测老龄化、治疗疾病(如阿尔茨海默氏病)和生产光学纯氨基酸都有着重要的作用.1-7缬氨酸是人体和其他许多生物所必需的8种氨基酸之一,如果缺乏会引起神经障碍、发育缓慢、消瘦和贫血等疾病.8-10D-缬氨酸是一种重要的有机手性源,广泛应用于生物、医学研究以及药物合成中,11-13但在自然界中缬氨酸主要以L型存在,探讨缬氨酸的旋光异构对控制L-缬氨酸的消旋现象和寻找D-缬氨酸的制备方法有重要的科学价值.
Dakin14在1910年提出的溶剂中氨基酸的消旋机理中指出,在最初的限速步骤中,L型氨基酸中α碳上的质子脱落,留下一个平面碳负离子后,围绕在周围的质子可以从两个方向攻击α碳,然后形成L型氨基酸或D型氨基酸.基于上述机理,Neuberger15推测,“高浓度的质子”更容易使游离的氨基酸消旋.这个机理表明两个质子是从两性离子的氨基上和次甲基碳上失去的.16按照Neuberger的方法, Smith和Sivakua17在1983年通过研究苯氨基乙酸消旋动力学证实了氨基酸的这种消旋机制.
对于单一的气相氨基酸分子,Sullivan等18基于量子化学计算提出了一个不同的反应机理,在其中,氢原子从手性碳上脱落,越过能垒,最后连接在氨基基团上形成中间体,而氨基基团上的另外一个氢原子迁移回到手性碳原子上,反应的能垒约为300 kJ·mol-1,由于位垒过高,这样的反应过程在温度较低的情况下很难发生.Wei等19,20指出在紫外线或阳光照射条件下L型氨基酸的晶体(L-Tyr)通过旋光异构反应可以得到D型氨基酸,但是插入层状双金属氢氧化物(水滑石)之间的L型氨基酸则不会出现这种现象,Sullivan等18提出的有关L型氨基酸发生旋光异构的理论模型无法解释此实验现象,而应用激发态理论则可以对涉及光反应的化学过程加以描述和解释.
缬氨酸作为含有五个碳原子的支链非极性α氨基酸,相比于绝大多数手性氨基酸具有简单的几何构型,而其相对于具有最简单的几何构型的丙氨酸在-R基上可以提供较明显的空间效应.因此本文利用量子化学方法对L-缬氨酸的旋光异构光反应机理进行了研究,并讨论了溶剂化效应对反应机理的影响有着特殊的意义.
2 计算方法
利用密度泛函理论(DFT)和从头算方法分别在B3LYP/6-311++G(d,p)以及MP2/6-311++G(d,p)水平上全优化基态(S0态)和第一激发三重态(T1态)的反应物(L-缬氨酸,即为L-VAL)、中间体(L-IM, D-IM)、产物(D-缬氨酸,即为D-VAL)以及过渡态(TSІ、TSII、TSIII)的几何构型,用含时密度泛函理论(TD-DFT)方法优化第一激发单重态(S1态)的反应物和产物的几何构型.用极化连续模型(PCM)方法进行溶剂化效应计算.全部计算采用Gaussian 09量子化学计算程序.21
3 结果与讨论
计算结果表明,L-VAL旋光异构反应包括两条可能反应路径,每一条途径都有三个反应步骤(如图1所示),即L-VAL首先由手性碳原子上的质子氢发生分子内迁移形成L-IM,然后L-IM通过单键旋转形成D-IM,D-IM再通过质子氢的迁移形成最终的D-VAL.反应途径以TSII为界具有对称性,反应的最终步骤可看作初始步骤的逆反应,D-IM与L-IM, D-VAL与L-VAL是具有镜面对称性的旋光异构体,其键长键角完全一致,二面角绝对值相同,数值相反.反应的初始步骤主要在于接受质子的中转媒介分别是羰基上的O和氨基上的N,下面将分别对其进行讨论.
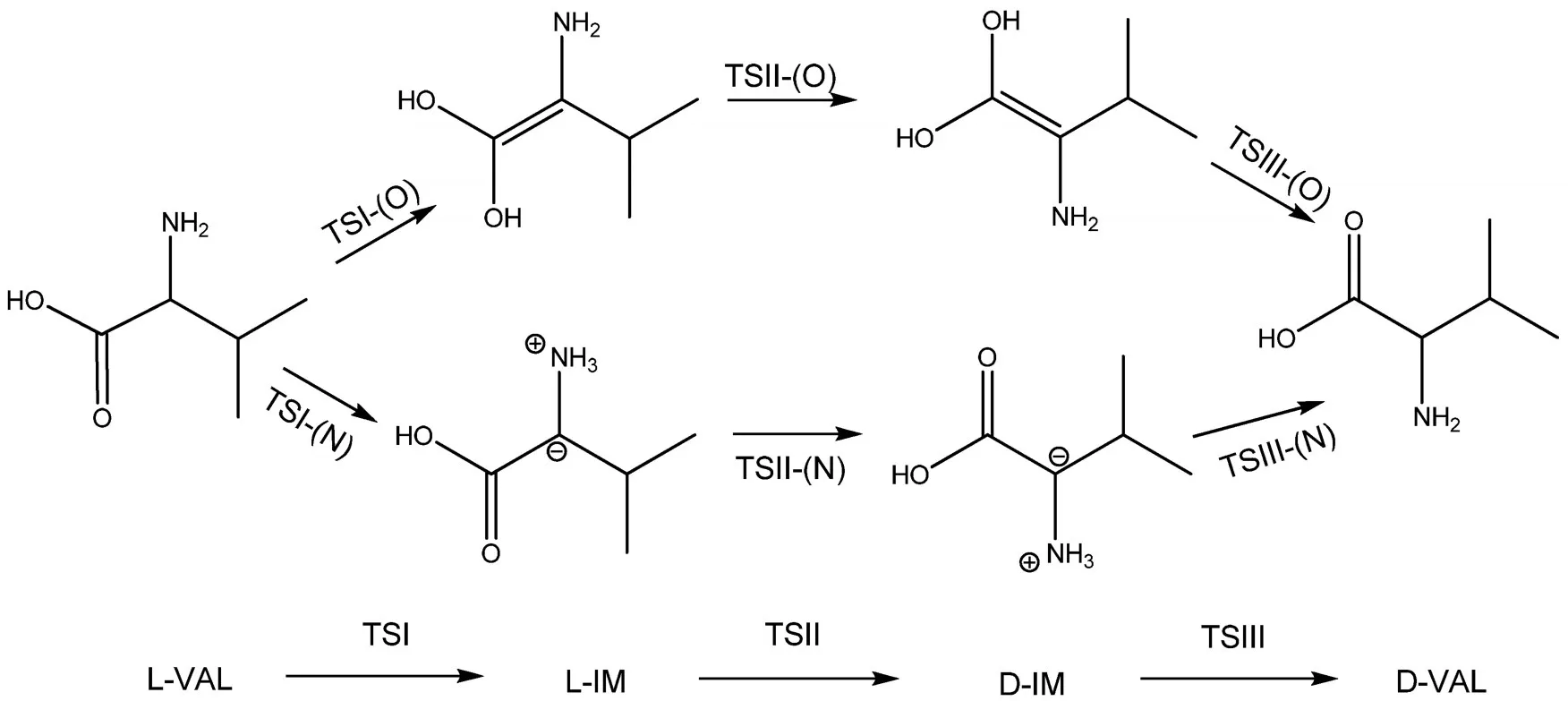
图1 L-缬氨酸旋光异构反应路径示意图Fig.1 Schematic diagram of the paths of L-valine optical isomerization
3.1 缬氨酸和中间体几何结构特征
L-VAL在S0、T1和S1态的结构模型如图2所示.基态气相缬氨酸分子的构象在文献中已有较多讨论,22本文计算得到的缬氨酸几何构型参数和振动频率与其结果基本一致,最显著的特征是分子内-COOH的H与-NH2的N的氢键相互作用,H和N之间的距离只有0.188 nm.在进行几何结构优化之前,曾反复设定L-VAL为两性离子形式的不同初始构型,即H与N为成键模式,但优化结果仍得到的是氢键形式,这表明缬氨酸分子的氢键形式比两性离子形式更稳定,这与Li等23在文献中所报道的结果相符.在T1态,L-VAL的C(3)-C(1),O(5)-C(3)和H(4)—C(1)的键长相对S0态都有所增加,二面角N(2)C(1)C(3)O(5)和O(6)C(3)C(1)O(5)的改变更为明显,尤其是后者从一个几乎是笔直的角度(-178.28°)扭转到-128.94°.在S1态,L-VAL的C(3)-C(1),O(5)-C(3)和H(4)-C(1)键长相对于S0态有所增加,但没有T1态明显,而二面角O(6)C(3)C(1)O(5)为-134.17°,也没有T1态扭转程度大.
用DFT方法在L-VAL旋光异构的两条反应路径上分别优化得到了S0和T1态的两种中间体,即以羰基氧为中转媒介反应路径上的烯醇式中间体(L-IM-(O),D-IM-(O))和以氨基氮为中转媒介反应路径上的两性离子中间体(L-IM-(N),D-IM-(N))(如图3所示).曾尝试用TD-DFT方法优化对应S1态的中间体的几何构型,但优化结果始终不收敛,故未确定S1态中间体构型.
在S0态,L-IM-(O)的C(3)-C(1)距离为0.134 nm,O(5)-C(3)的键长为0.137 nm,近似于O(6)-C(3)的键长(0.138 nm),同样H(4)-O(5)与H(7)-O(6)键长都约为0.096 nm,二面角N(2)C(1)C(3)O(5)几乎是0°,这表明N(2)、C(1)、C(3)、C(10)、O(5)和O(6)在同一平面,C(1)已不再是手性C原子,并可以从两个方向接受迁移的质子氢.在T1态,L-IM-(O)的几何构型变化非常明显,N(2)、C(1)、C(3)、C(10)、O(5)和O(6)原子已不在同一平面,C(1)-N(2)键几乎垂直于C(3)、O(5)和O(6)所在的平面,二面角N(2)C(1)C(3)O(5)从基态的11.25°扭转到69.79°.
在S0态,L-IM-(N)的C(3)-C(1)距离为0.139 nm,H(4)-N(2)的键长与H(8)-N(2),H(9)-N(2)的键长几乎相同(0.103 nm),二面角C(3)C(1)N(2) H(4)为-118.44°,二面角C(3)C(1)N(2)C(10)几乎是近似为180°,这意味着N(2)、C(1)、C(3)、C(10)在同一平面,C(1)已不再是手性C原子,并可以从两个方向接受迁移的质子氢.在T1态,L-IM-(N)的几何构型相对于基态的变化并不十分明显.
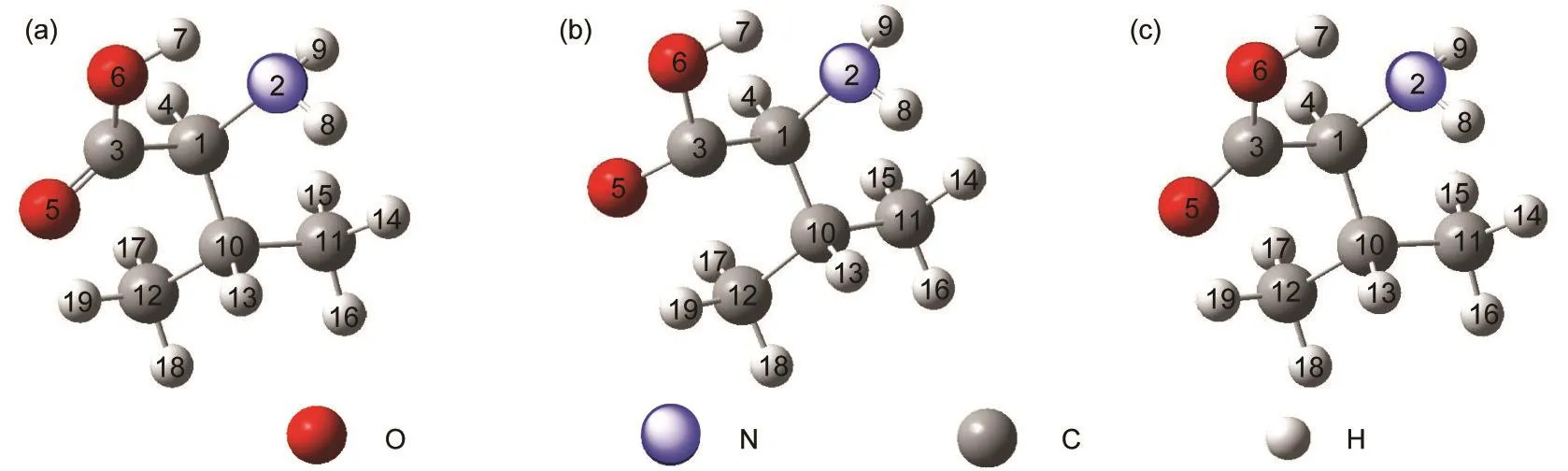
图2 L-缬氨酸在不同态的几何结构模型Fig.2 Structural models of L-valine on different states (a)S0state;(b)T1state;(c)S1state
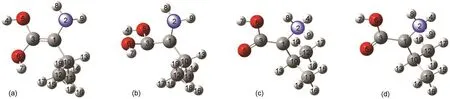
图3 两种中间体在不同态下的几何结构模型Fig.3 Structural models of two kinds of intermediates on different states(a)L-IM-(O)on S0state;(b)L-IM-(O)on T1state;(c)L-IM-(N)on S0state;(d)L-IM-(N)on T1state
3.2 S0和T1态过渡态的结构特征
在L-VAL的旋光异构反应中,从L-VAL到L-IM是手性碳原子上的质子氢发生分子内迁移的过程,质子氢的迁移有两条可能的路径.下面对这两条质子迁移路径上的过渡态分别进行说明.
如图4所示,第一条反应路径上,在S0态,过渡态TSI-(O)的C(3)-C(1)键长从L-VAL的0.155 nm下降到0.144 nm,O(5)-C(3)的键长从0.120 nm增加到0.129 nm,最为显著的变化是C(1)-H(4)的距离从0.110 nm变为0.158 nm,相应的O(5)-H(4)的距离从0.288 nm缩小到0.123 nm,角H(4)-C(1)-C(3)从104.76°减小到63.43°,另一个显著的结构变化是二面角N(2)C(1)C(3)O(5)沿着C(1)-C(3)的方向的扭转了约55°,使羧基O(5)位于一个有利的位置来接受迁移的质子.在T1态,TSI-(O)键长和二面角的变化与基态的变化趋势一致,相对于基态键长O(5)-C(3),O(6)-C(3)和C(3)-C(1)分别有所增加,二面角N(2)C(1)C(3)O(5)则减小.第二条反应路径上在S0态,过渡态TSI-(N)中的C(3)-C(1)的键长从反应物的0.155 nm下降到0.142 nm,N(2)-C(1)的键长从0.148 nm增加到0.159 nm,最为显著的变化是C(1)-H(4)的距离从0.110 nm变为0.137 nm,相应的N(2)-H(4)的距离从0.207 nm缩小到0.117 nm,角H(4)-C(1)-N(2)从106.19°减小到45.81°,另一个显著的结构变化是二面角C(3)C(1)N(2)H(8)沿着C(1)-N(2)的方向的扭转了约75°,使氨基上的N(2)位于一个有利的位置来接受迁移的质子.在T1态,TSI-(N)键长和二面角的变化与基态的变化趋势一致,结构性变化并不明显.
旋光异构反应过程中若中转媒介上的质子氢要从另一方向迁移回手性C(1)原子上,L-IM必先通过TSII转变成D-IM.第一条路径上过渡态TSII-(O)主要变化是二面角N(2)C(1)C(3)O(5)沿着C(1)-C(3)的方向扭转,使得质子氢可以在接下来的反应中处于更有利的迁移位置.第二条路径上过渡态TSII-(N)主要表现的是二面角C(3)C(1)N(2)H(8)沿着C(1)-N(2)的方向扭转,使得质子H可以在接下来的反应中处于更有利的迁移位置.
D-IM通过TSIII最终转变为D-VAL,其过程可看作是从L-VAL到L-IM反应的逆过程,中转媒介上的质子氢发生分子内迁移向手性C原子靠近并最终与之形成C-H键.在计算的TSIII和TSI的结构参数中,键长与键角均对应相等,二面角绝对值相同,符号相反.
3.3 L-VAL的旋光异构光反应机理
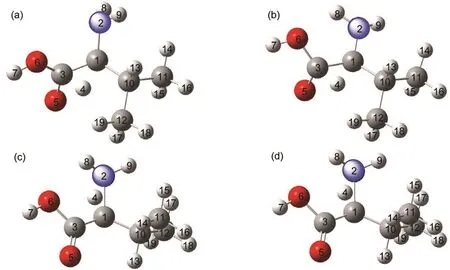
图4 两种过渡态(TSI)在不同态下的几何结构模型Fig.4 Structural models of two kinds of transition state(TSI)on different states (a)TSI-(O)on S0state;(b)TSI-(O)on T1state;(c)TSI-(N)on S0state;(d)TSI-(N)on T1state
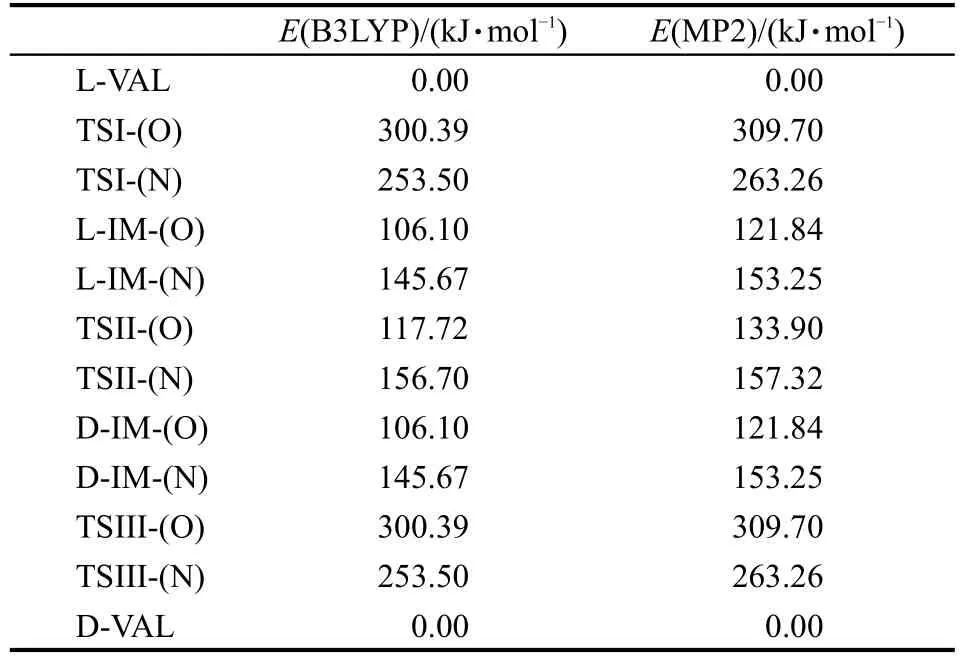
表1 利用不同方法和6-311++G(d,p)基组计算的包含零点能校正的S0态各物种相对能量Table 1 Relative energies including zero-point energy corrections computed by different methods and 6-311++G(d,p)basis set on S0state
根据反应过程中各物种的能量可以确定反应的能垒,为了直观起见,表1给出了包含S0态零点能校正的以L-VAL能量为基准,分别用B3LYP/6-311++G(d,p)以及MP2/6-311++G(d,p)方法计算得到的各物种的相对能量.计算数据表明密度泛函和从头算方法对反应能垒的影响相差较小,不超过10 kJ·mol-1,这表明B3LYP与MP2方法均适合于研究L-VAL旋光异构反应路径上各物种的分子结构.由于L-VAL旋光异构光反应途径为对称途径,反应途径上L-VAL与D-VAL、L-IM与D-IM、TSI与TSIII的能量均对应相等.根据密度泛函方法计算得到的L-VAL旋光异构反应能量变化如图5所示,在S0态下,沿着两条氢迁移路径,其中每条反应路径上都需要跨过三个能垒,第一条路径上的能垒分别为300.39、11.62和194.29 kJ·mol-1;第二条路径上的能垒分别为253.50、11.03和107.83 kJ·mol-1.其中第一个和第三个能垒即反应通过TSI和TSIII时的能垒很高,因此缬氨酸在S0态发生旋光异构的可能性很小,尤其是缬氨酸形成中间体的第一个能垒达到300 kJ·mol-1以上,这正是氨基酸在常温下很难发生消旋现象的原因.
通常光反应机理都极为复杂,结构性的变化决定了各态能量的变化,在T1态时反应过程的能垒下降十分明显.如图5所示,第一条路径上L-VAL经过TSI-(O)到L-IM-(O),在基态时需要跃过300.39 kJ· mol-1的能垒,反应很难发生,因此,消旋现象的实验研究都是在加热或光照条件下进行的.19根据Frank-Condon原理以及计算结果可知,S0态L-VAL在紫外光照射下首先被垂直激发到S1态,然后弛豫到S1态的平衡态,能量为424.58 kJ·mol-1,由于S1态的反应位垒一般较低,L-VAL有可能通过S1态发生旋光异构反应,最后从S1态的D-VAL经过磷光过程回到S0态并完成最终的反应,但是S1态的寿命较短,在S1态进行的反应过程中极容易经过系间窜跃到达T1态,反应完全在S1态进行的可能性不大,由于未能优化出S1态上的L-IM、D-IM结构,所以在此不做深入的讨论.另一种可能途径是S1态L-VAL经过系间窜跃形成T1态L-VAL,然后在T1态克服55.83 kJ· mol-1的能垒经过TSI-(O)到L-IM-(O),此反应过程应该容易完成.L-IM-(O)有可能从T1态回落到S0态,虽然L-IM-(O)到D-IM-(O)只不过是经过了单键旋转,反应能垒很低,反应极易进行,但是在S0态D-IM-(O)到D-VAL需要跨过194.29 kJ·mol-1的能垒,此过程并不十分容易发生,所以T1态L-IM-(O)也可能直接通过单键旋转变成D-IM-(O)后跃过126.16 kJ·mol-1的能垒直接到达T1态的D-VAL,然后通过发射磷光落回到S0态完成最终的反应.以氨基N为中转媒介的缬氨酸旋光异构反应途径整体上与此相似.
3.4 溶剂化效应对该反应的影响
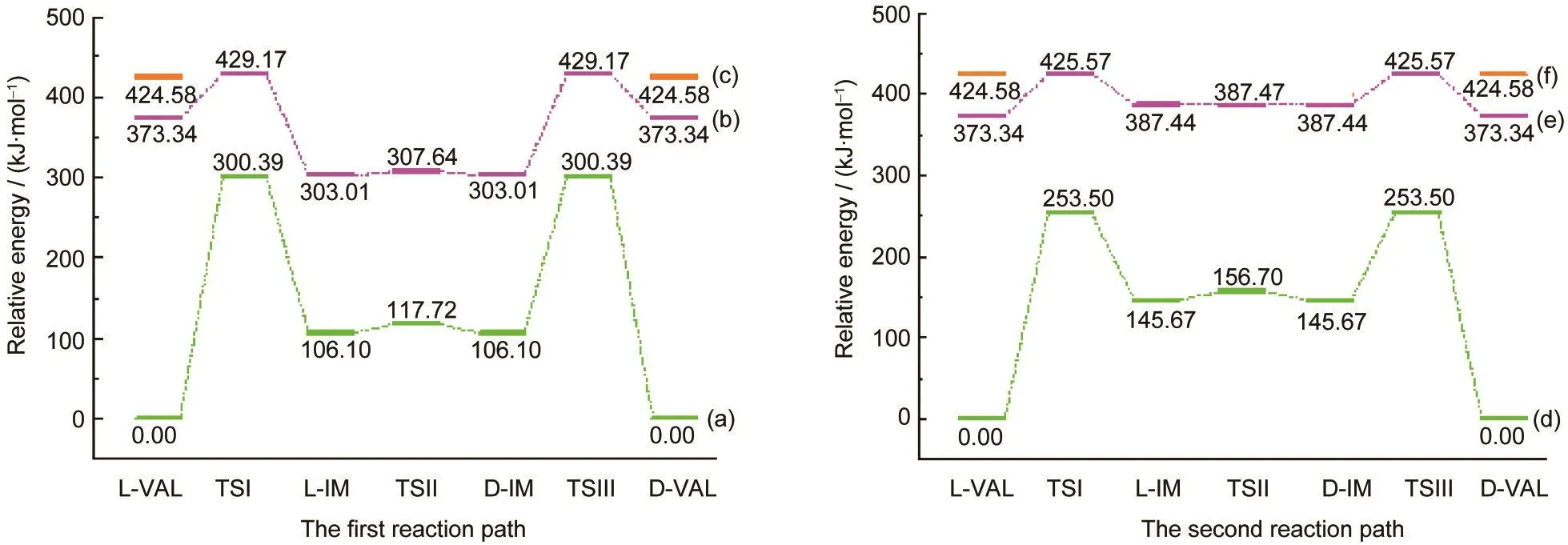
图5 L-缬氨酸旋光异构反应在两条反应途径上的能量变化Fig.5 Energy variations of L-valine optical isomerization on two reaction paths (a,d)S0state;(b,e)T1state;(c,f)S1state
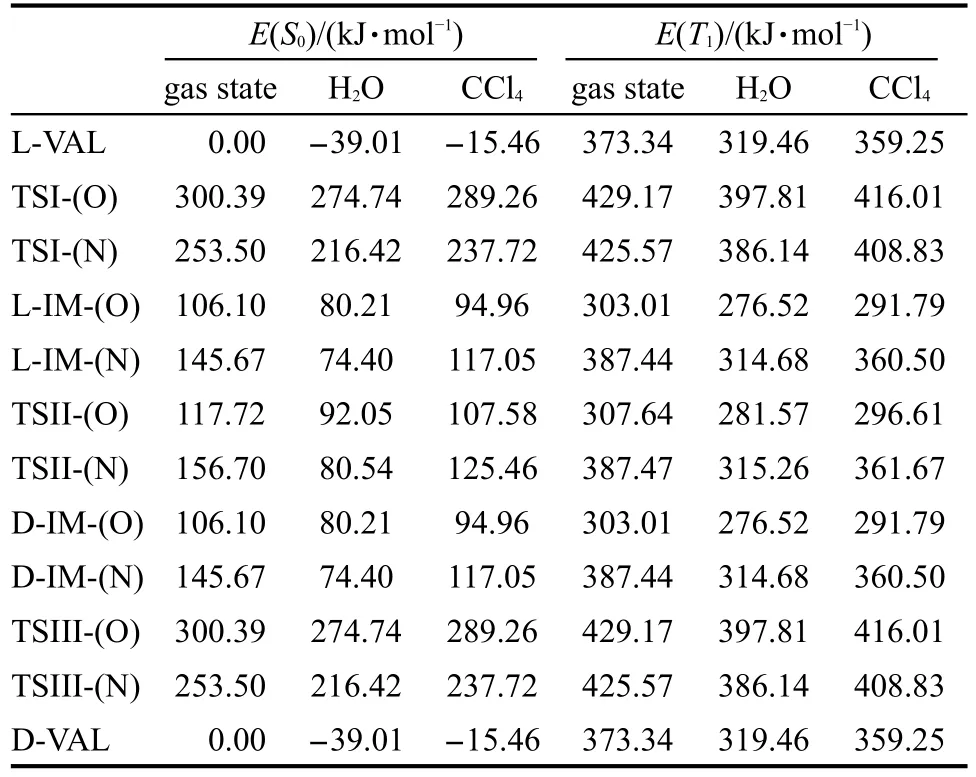
表2 在B3LYP/6-311++G(d,p)水平上得到的溶剂化效应对L-缬氨酸旋光异构反应各物种相对能量的影响Table 2 Influences of solvent effect on the relative energies of all species of L-valine optical isomerization at the level of B3LYP/6-311++G(d,p)
众所周知,缬氨酸在水溶液中以电离的形式进行旋光异构反应,但是缬氨酸在非极性溶剂中或者蛋白质中的缬氨酸残基在水溶液中是无法电离的,在这种情况下缬氨酸的旋光异构反应与气态时的形式一样,那么此时溶剂对此旋光异构反应就存在一定的影响.24-26因此对上述反应要进行更深一步的探讨.考虑到极性与非极性以及无机溶剂与有机溶剂等因素对旋光异构反应的影响,选择无机极性溶剂水(H2O)与有机非极性溶剂四氯化碳(CCl4)作为溶剂对该反应进行溶剂化效应计算.表2给出了在B3LYP/6-311++G(d,p)水平上计算得到的已包含零点能校正的溶剂化效应对L-缬氨酸旋光异构反应各物种相对能量的影响,观察能量变化可知CCl4的溶剂化效应对L-VAL的旋光异构反应影响不大, H2O的溶剂化效应对第一条反应途径的影响同样很小,但对第二条反应途径有明显的影响,尤其是第三个反应能垒相差了约30 kJ·mol-1.这可能是由于L-IM-(N)是两性离子,H2O中的质子H易与L-IM-(N)中的C负离子形成氢键,从而对分子内质子H迁移回手性C上形成阻碍,导致能垒偏高.溶剂化效应对旋光异构反应途径上各物质能量都有降低趋势的影响,无机极性溶剂(H2O)对能量影响相对有机非极性溶剂(CCl4)更大一些.对比基态与激发态反应途径上各物质能量变化可知溶剂化效应对其影响同样不大.
4 结论
利用B3LYP和MP2方法在6-311++G(d,p)的计算水平上对L-VAL旋光异构反应进行了研究,通过分析反应物、中间体和产物的结构变化发现了分别以羰基氧原子和氨基氮原子为中转媒介的两条可能的质子迁移反应路径.
分析L-VAL在S0、T1和S1态反应过程中能量的变化,可以推测L-VAL的旋光异构光反应机理.由于S0态反应位垒较高,在基态发生旋光异构反应的可能性很小.在光反应条件下,紫外光首先使LVAL激发到S1态,并弛豫到局部稳定点.由于S1态的反应位垒一般较低,有可能通过S1态发生旋光异构反应,但通常S1态的寿命较短,反应完全在S1态进行的可能性不大.反应过程中随时有可能从S1态经过系间窜跃到达T1态.在光激发后,S1态L-VAL也可以经过系间窜跃到达T1态L-VAL,然后发生手性碳质子氢的分子内迁移,跃过能垒,连接在中转媒介羰基氧原子或氨基氮原子上形成T1态的L-IM,并通过单键旋转转变为T1态的D-IM.连接在中转媒介上的质子氢再次进行分子内迁移,从另一侧与手性碳相连,反应经过磷光过程,进入S0态并最终生成产物D-VAL.
通过溶剂化效应的计算可以看出,CCl4的溶剂化效应对L-VAL旋光异构反应的影响较小,H2O的溶剂化效应对第二条反应途径有明显的阻碍作用.溶剂化效应对旋光异构反应途径上各物质能量都有降低趋势的影响,对基态与激发态反应途径上各物质能量变化影响不大,无机极性溶剂对能量的影响相对有机非极性溶剂更大一些.
(1)Minami,M.;Takeyama,M.;Mimura,K.;Nakamura,T.Nucl. Instrum.Methods Phys.Res.Sect.B 2007,259,547.doi: 10.1016/j.nimb.2007.01.201
(2)Amelung,W.;Zhang,X.;Flach,K.W.Geoderma 2006,130, 207.doi:10.1016/j.geoderma.2005.01.017
(3) Sajdok,J.;Kozak,A.;Zidkova,J.;Kotsba,P.;Pilin,A.;Kas,J. Chem.Listy 2001,95,98.
(4)Tomiyama,T.;Asano,S.;Furiya,Y.;Shirasawa,T.;Endo,N.; Mori,H.J.Biol.Chem.1994,269,10205.
(5) Friedman,M.J.Agric.Food Chem.1999,47,3457.doi: 10.1021/jf990080u
(6) Martineau,M.;Baux,G.;Mothet,J.P.J.Physiol.-Paris 2006, 99,103.doi:10.1016/j.jphysparis.2005.12.011
(7) Chu,Y.Q.;Pan,T.T.;Dai,Z.Y.;Yu,Z.W.;Zheng,S.B.;Ding, C.F.Acta Phys.-Chim.Sin.2008,24,1981. [储艳秋,潘婷婷,戴兆云,俞卓伟,郑松柏,丁传凡.物理化学学报,2008,24, 1981.]doi:10.3866/PKU.WHXB20081108
(8) Qi,J.Studies on Configuration Transformation of L-Proline and L-Valine.M.S.Dissertation,Nanchang University,Jiangxi, 2006.[漆 剑.L-脯氨酸和L-缬氨酸构型转换的研究[D].南昌:南昌大学,2006.]
(9) Mei,L.H.;Yao,S.J.;Guan,Y.X.;Lin,D.Q.Chinese Journal of Pharmaceuticals 1999,30(5),235.[梅乐和,姚善泾,关怡新,林东强.中国医药工业杂志,1999,30(5),235.]
(10)Chen,Y.;Wang,W.Q.;Du,W.M.Acta Phys.-Chim.Sin.2004, 20,540. [陈 渝,王文清,杜为民.物理化学学报,2004,20, 540.]doi:10.3866/PKU.WHXB20040519
(11) Frauli,M.;Ludwig,H.Arch.Gynecol.Obstet.1987,241,87. doi:10.1007/BF00931229
(12)Picciano,P.T.;Johnson,B.;Walenga,R.W.;Donovan,M.; Douglas,B.J.;Kreutzer,D.L.Exp.Cell Res.1984,51,134.
(13) Li,A.P.;Zhao,Q.;Cheng,X.C.;Xu,H.Q.Journal of Anhui Agricultural Sciences 2010,38(14),7208. [李爱平,赵 青,程晓春,徐海青.安徽农业科学,2010,38(14),7208.]
(14)Dakin,H.D.Am.Chem.J.1910,44,48.
(15) Neuberger,A.Adv.Protein Chem.1948,4,297.doi:10.1016/ S0065-3233(08)60009-1
(16) Ebbers,E.J.;Ariaans,G.J.A.;Houbiers,J.P.M.;Bruggink,A.; Zwanenburg,B.Tetrahedron 1997,53,9417.doi:10.1016/ S0040-4020(97)00324-4
(17) Smith,G.G.;Sivakua,T.J.Org.Chem.1983,48,627.
(18) Sullivan,R.;Pyda,M.;Pak,J.;Wunderlich,B.;Thompson,J. R.;Pagni,R.;Pan,H.;Barnes,C.;Schwerdtfeger,P.;Compton, R.J.Phys.Chem.A 2003,107,6674.doi:10.1021/jp0225673
(19)Wei,M.;Xu,X.Y.;He,J.;Yuan,Q.;Rao,G.Y.;Evans,D.G.; Pu,M.;Yang,L.J.Phys.Chem.Solids 2006,67,1469.doi: 10.1016/j.jpcs.2006.01.118
(20) Wei,M.;Pu,M.;Guo,J.;Han,J.B.;Li,F.;He,J.;Evans,D.G.; Duan,X.Chem.Mater.2008,20,5169.doi:10.1021/ cm800035k
(21) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, RevisionA.01;Gaussian Inc.:Wallingford,CT,2009.
(22) Linder,R.;Nispel,M.;Haber,T.;Kleiermanns,K.Chem.Phys. Lett.2005,409,260.doi:10.1016/j.cplett.2005.04.109
(23) Li,J.;Brill,T.B.J.Phys.Chem.A 2003,107,5993.doi: 10.1021/jp022477y
(24) Gomzi,V.;Herak,J.J.Mol.Struct.-Theochem 2003,629,71.
(25) Ren,X.H.Theoretical Study of Interaction between Solvent andAminoAcids.M.S.Dissertation,Jiangnan University, Jiangsu,2008.[任晓慧.氨基酸分子与溶剂间相互作用的理论研究[D].无锡:江南大学,2008.]
(26)Yu,Y.;Huang,D.F.;Wang,D.X.Chin.J.Chem.Phys.2005, 18,336. [俞 英,黄东枫,王大喜.化学物理学报,2005,18, 336.]
March 27,2012;Revised:May 15,2012;Published on Web:May 16,2012.
Two Possible Photoreaction Pathways of L-Valine Optical Isomerization
MAChi-Cheng1PU Min1,*WEI Min1LI Jun-Nan1LI Zhi-Hong2
(1State Key Laboratory of Chemical Resource Engineering,Beijing University of Chemical Technology,Beijing 100029,P.R.China;2BeijingSynchrotronRadiationFacility,InstituteofHighEnergyPhysics,ChineseAcademyofSciences,Beijing100049,P.R.China)
The photoreaction mechanism of L-valine optical isomerization is studied by density functional theory(DFT)and ab initio molecular orbital theory.The geometric parameters of reactant,product, intermediates,and transition states,as well as the reaction energy barriers,on the reaction paths in S0and T1states are optimized at the B3LYP and MP2 levels using the 6-311++G(d,p)basis set.The equilibrium geometries of the S1state of valine are optimized using time dependent density functional theory(TD-DFT)at the B3LYP/6-311++G(d,p)level.From the analysis of each geometric stationary point on the reaction path, the photoreaction mechanism of L-valine optical isomerization is proposed.The reaction proceeds via hydrogen transfer with the help of carbonyl O or amino N atom in the excited state.Furthermore,the effect of solvent on the isomer reaction mechanism is discussed based on the polarizable continuum model (PCM)of self-consistent reaction field theory.
Valine;Optical isomerization;Density functional theory;Ab initio molecular orbital theory; Solvent effect
10.3866/PKU.WHXB201205162
O641
∗Corresponding author.Email:pumin@mail.buct.edu.cn;Tel:+86-10-64443922.
The project was supported by the National Natural Science Foundation of China(21173019,11079041).
国家自然科学基金(21173019,11079041)资助项目
