Co(II)与烟酸类分子形成的配位聚合物的晶体结构与孔道稳定性
2012-12-21华小辉李维红徐怡庄吴瑾光
华小辉 李维红 徐怡庄 吴瑾光
(北京大学化学与分子工程学院,北京100871)
Co(II)与烟酸类分子形成的配位聚合物的晶体结构与孔道稳定性
华小辉 李维红*徐怡庄 吴瑾光
(北京大学化学与分子工程学院,北京100871)
利用烟酸与异烟酸两种配体分别与硝酸钴在N,Nʹ-二甲基甲酰胺(DMF)中采用溶剂热法合成了三种新的配位聚合物[Co2(μ2-H2O)(nicotinic acid)4·(DMF)](1),[Co2(isonicotinic acid)4·(DMF)](2),[Co(isonicotinic acid)2·(DMF)](3),并利用单晶X射线衍射(XRD)和元素分析获得其结构信息.通过傅里叶变换红外(FTIR)光谱、粉末X射线衍射(XRD)、热重分析(TGA)和比表面积分析等手段对结构中孔道的热稳定性进行了表征.结果表明化合物1具有类金刚石的拓扑结构,结构中含有稳定的一维(1D)孔道空间.化合物2和3是以异烟酸为配体,分别在100和160°C下合成,二者结构中配体以完全不同的方式与Co(II)配位,从而使其具有不同的结构.化合物2和3的一维孔道在热处理脱附DMF分子的过程中不能稳定存在.
配位聚合物; 烟酸;晶体结构;拓扑;傅里叶变换红外光谱;孔道稳定性
1 引言
配位聚合物因其丰富的结构特点1-4和由此带来的光学、磁学、催化、气体吸附等方面的性质,5-16一直以来都是人们重点研究的领域.目前倍受人们关注的金属-有机骨架材料即是利用有机分子与金属配位形成的具有孔道结构的一类配位聚合物.17-21通常情况下,含有羧酸或吡啶基团的有机分子是人们优先选择的配体.羧酸类配体由于与金属具有较强的配位能力和多样的配位方式,经常被人们所采用.22如Yaghi等17,23,24曾系统地研究过对苯二甲酸与Zn2+、Cu2+、Co2+、Mn2+、Tb3+等金属离子的配位情况,合成了一系列结构多样的孔道材料,其中著名的MOF-5即是利用Zn2+与对苯二甲酸作用形成的. 4,4ʹ-联吡啶作为一个中性的桥连配体,它与Zn2+、Cd2+等金属离子配位形成的多孔结构也被人们广泛研究.25-27羧酸和吡啶配体的混合使用在文献28-30中也屡见报道.除此之外,多官能团配体中多个官能团的协同配位也会带来丰富多样的孔道结构.Xiang等31利用同时含有吡啶和羧酸根的配体与Cu2+作用,合成的两种配合物均含有纳米尺度的孔道并具有较高的比表面积.Eubank等32通过化学反应在间苯二甲酸上连接了吡啶基团,所合成的配体与Cu2+作用得到一系列具有丰富拓扑网络和孔道结构的化合物.本文利用烟酸和异烟酸两种同时含有羧酸和吡啶基团的配体与Co(II)作用,考察不同空间位置下羧酸与吡啶基团的协同配位情况,并考察溶剂热反应温度对配合物结构的影响.
2 实验部分
2.1 试剂和仪器
烟酸、异烟酸、Co(NO3)2·6H2O为国药集团化学试剂公司分析纯试剂,N,Nʹ-二甲基甲酰胺(DMF)从TCI公司购得,纯度>99%,使用前未做进一步处理.样品中C、H、N含量的测定是在德国的Elementar Analysensysteme GmbH生产的vario EL型元素分析仪上完成.显微红外光谱采用美国Thermo Fisher公司生产的Nicolet iN10显微红外仪测定.热重曲线是在美国TA公司的Q600热重-差热同步测定仪上测定,N2气氛,升温速率为10°C·min-1.粉末X射线衍射数据是在日本Rigaku D/max-2000转靶X射线衍射仪上收集,石墨单色器,Cu Kα射线(λ=0.15418 nm),扫描范围3°-40°,步长0.02°,管电流100 mA,管电压40 kV.N2吸附-脱附曲线是在美国Micrometer公司生产的ASAP2010比表面积测定仪上测定,测定温度77 K.
2.2 配合物的合成
2.2.1 Co2(μ2-H2O)(nicotinic acid)4·(DMF)(1)的合成
称取0.024 g(0.2 mmol)的烟酸和0.028 g(0.1 mmol)的Co(NO3)2·6H2O加入5 mL的烧杯中,向烧杯中加入2 mL的DMF后超声使固体溶解,再将烧杯置于20 mL的水热釜中,密封后130°C加热24 h,得到粉色的长条状晶体,产率83%.元素分析显示其组成为C27H25Co2N5O10(理论值(%):C 46.24,H 3.66,N 10.14;实验值(%):C 46.50,H 3.61,N 10.14).
2.2.2 Co2(isonicotinic acid)4·(DMF)(2)的合成
称取0.051 g(0.4 mmol)的异烟酸和0.060 g (0.2 mmol)的Co(NO3)2·6H2O加入10 mL的烧杯中,向烧杯中加入4 mL的DMF,再将烧杯置于20 mL的水热釜中,密封后100°C加热24 h,得到大量粉色的块状晶体和少量的紫色长条状晶体,用DMF洗涤三次除去溶液中灰色的悬浊物,粉色晶体的产率为70%.元素分析显示其组成为C27H23Co2N5O9(理论值(%):C 47.33,H 3.41,N 10.30,实验值(%):C 44.60, H 3.49,N 9.88).
2.2.3 Co(isonicotinic acid)2·(DMF)(3)的合成
称取0.051 g(0.4 mmol)的异烟酸和0.060 g (0.2 mmol)的Co(NO3)2·6H2O加入10 mL的烧杯中,向烧杯中加入4 mL的DMF,再将烧杯置于20 mL的水热釜中,密封后160°C加热24 h,得到紫色长条状晶体,产率为88%.元素分析显示其组成为C15H15CoN3O5(理论值(%):C 47.86,H 4.01,N 11.17;实验值(%):C 46.02,H 4.40,N 11.40).
2.3 晶体结构的测定
单晶X射线衍射数据是在日本Rigaku Saturn 724单晶衍射仪上收集的,检测器为Saturn 724+CCD面探测器,石墨单色器,Mo Kα射线(λ= 0.071073 nm).晶体结构采用SHEXS-97程序的直接法解出,33并采用SHEXL-97程序的全矩阵最小二乘法精修.34非氢原子均进行各向异性精修.配体上的氢原子均采用理论加氢的方法固定在C原子上(C―H键长0.093 nm),水分子上的氢利用差值傅里叶图求得.化合物1含有无序的DMF分子,常规的手段很难得到满意的结果,采用platon软件中的Squeeze程序去除无序分子引起的电荷密度.35化合物1的化学式是结合晶体数据与热重、元素分析等结果计算出来的.化合物1、2、3的晶体学数据见表1,该数据已存于英国剑桥晶体学数据中心(CCDC 886303、886304、886305).
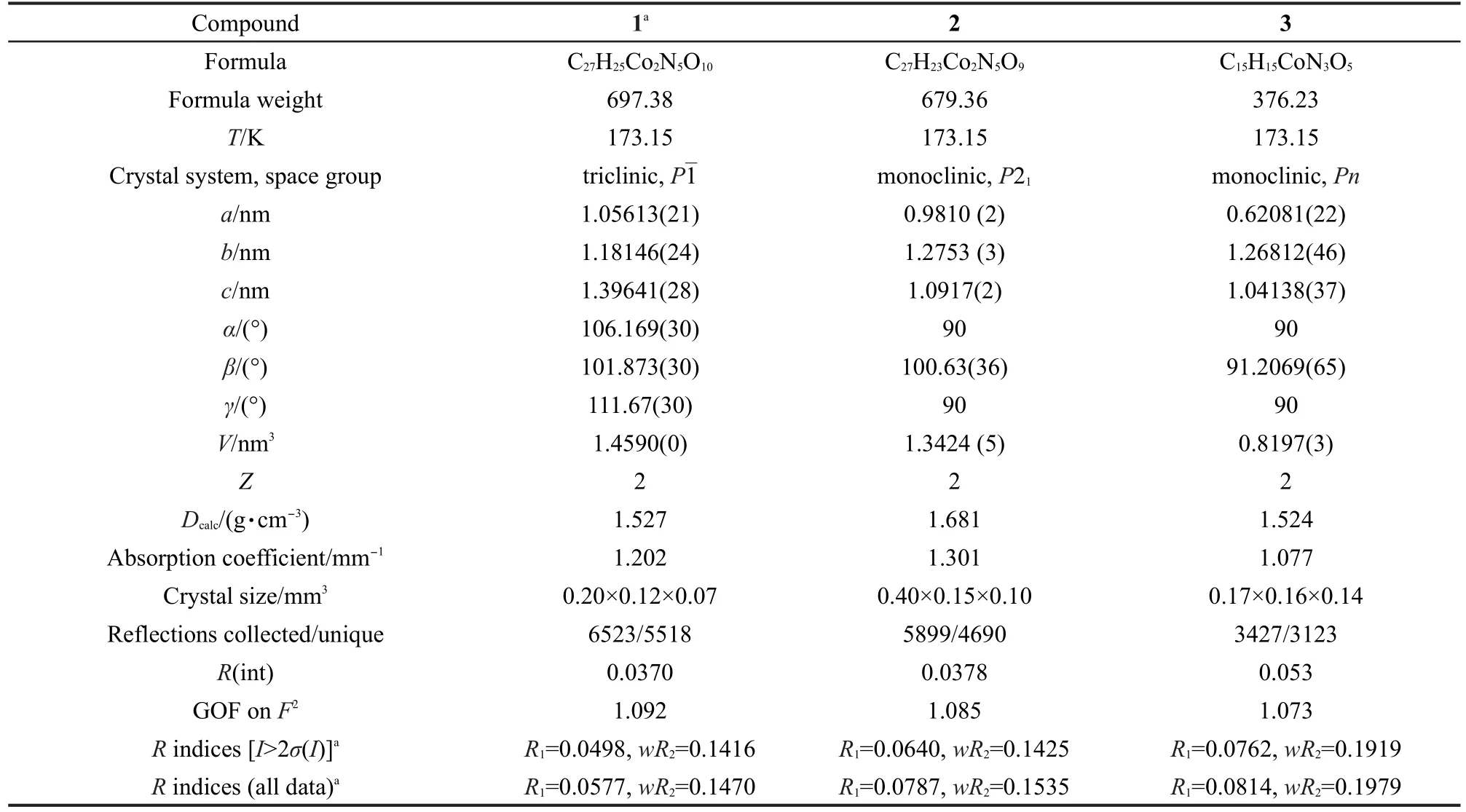
表1 化合物1-3的晶体学参数Table 1 Crystallographic parameters for compounds 1-3
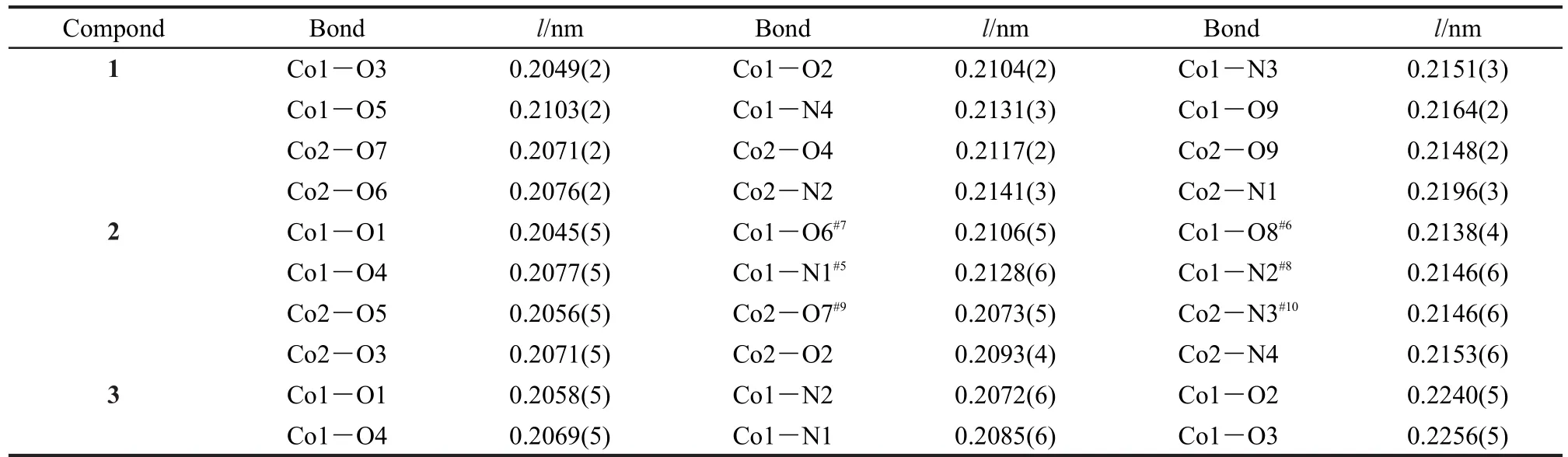
表2 化合物1-3的部分键长(l)Table 2 Selected bond lengths(l)for compounds 1-3
3 结果与讨论
3.1 晶体结构
化合物1的结构属于三斜晶系P1空间群,其晶体结构如图1所示.化合物中含有Co(II)形成的1双核结构,除了起桥连作用的水分子外(l(Co1―O9)= 0.2164 nm,l(Co2―O9)=0.2148 nm,θ(Co2O9Co1)= 113.57°),Co1和Co2之间还依靠两个羧酸根桥连起来(l(Co1―O3)=0.2049 nm,l(Co2―O4)=0.2117 nm, l(Co1―O5)=0.2143 nm,l(Co2―O6)=0.2076 nm)(化合物1的主要键长见表2).两个金属中心Co1和Co2均采取六配位形式与配体作用,除配位水外,它们分别与来自不同配体的两个吡啶N及三个羧酸根的O配位.配体中的羧酸根共有两种配位模式:单齿配位(μ1-η1:η0)和双齿配位μ2-η1:η1.如图2(a)所示, Co2(μ2-H2O)可视作该结构的节点,这些节点在空间中利用八个配体与周围的其它四个节点相连.这些节点之间以四面体的形式连接起来(图2(b)),夹角为110°-137°.化合物1沿a轴方向形成一维的孔道结构,孔道尺寸约为0.4 nm×0.4 nm,见图2(c).该化合物在空间上的拓扑结构为三维(3D)的66的类金刚石型(diamond-like)网络,如图2(d)所示.这一骨架与Lin36和 Liu37等报道的[Co2(nicotinate)4(μ-H2O)]· CH3CH2OH·H2O基本类似.但文献36,37中采用的3-氰基吡啶在水和乙醇混合溶剂中以水解的方式与Co(II)作用,本文直接采用烟酸配体与Co(II)在DMF中反应制得,更为简便.文献中孔道中的分子是乙醇和水分子,而本文中是溶剂DMF分子,Co(II)与DMF的摩尔比为2:1.利用platon计算化合物1中的孔隙率,溶剂分子占据23.7%的空间.35
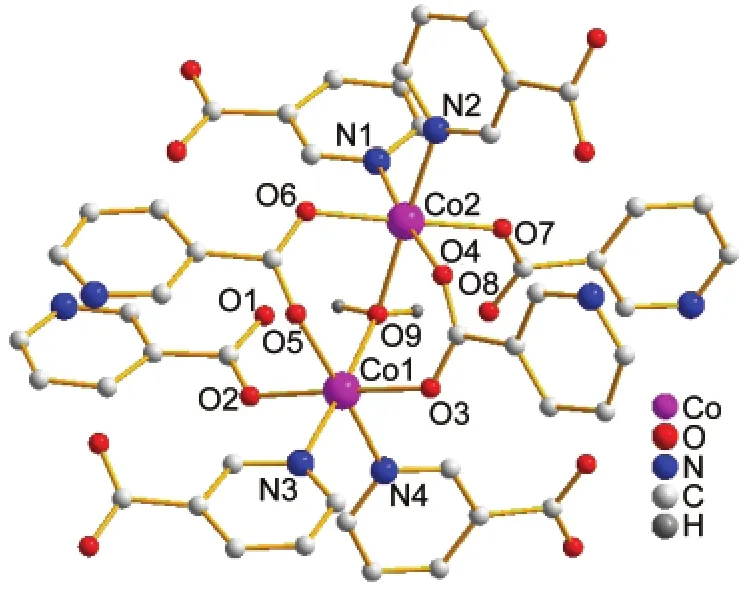
图1 化合物1中Co(II)的配位环境Fig.1 Coordination environment of Co(II)in compound 1Disordered DMF molecules and hydrogen atoms on ligands have been omitted for clarity.
化合物2则是利用异烟酸与Co(NO3)2·6H2O于100°C在DMF中反应制得,其结构属于单斜的P21空间群.如图3所示,结构中含有两个晶体学独立的Co(II),Co1和Co2具有相似的配位环境,Co1在赤道面上与四个羧酸根配位(l(Co1―O1)=0.2045 nm, l(Co1―O4)=0.2047 nm,l(Co1―O6)=0.2106 nm, l(Co1―O8)=0.2138 nm),而在轴向位置与两个吡啶N相连(l(Co1―N1)=0.2128 nm,l(Co1―N2)=0.2146 nm)(见表2).相邻的Co1与Co2通过两个羧酸根以μ2-η1:η1形式连接起来l(Co1···Co2=0.4863 nm),沿着a轴方向形成一维的链状结构(图4(a)).链之间通过Co(II)与吡啶基团的配位连成三维的网络结构,见图4(b),该化合物沿a轴方向形成一维的孔道结构,孔道中含有DMF溶剂分子,整个结构中Co(II)与DMF分子的摩尔比为2:1.同样利用platon计算,溶剂分子占据22.4%的空间.35
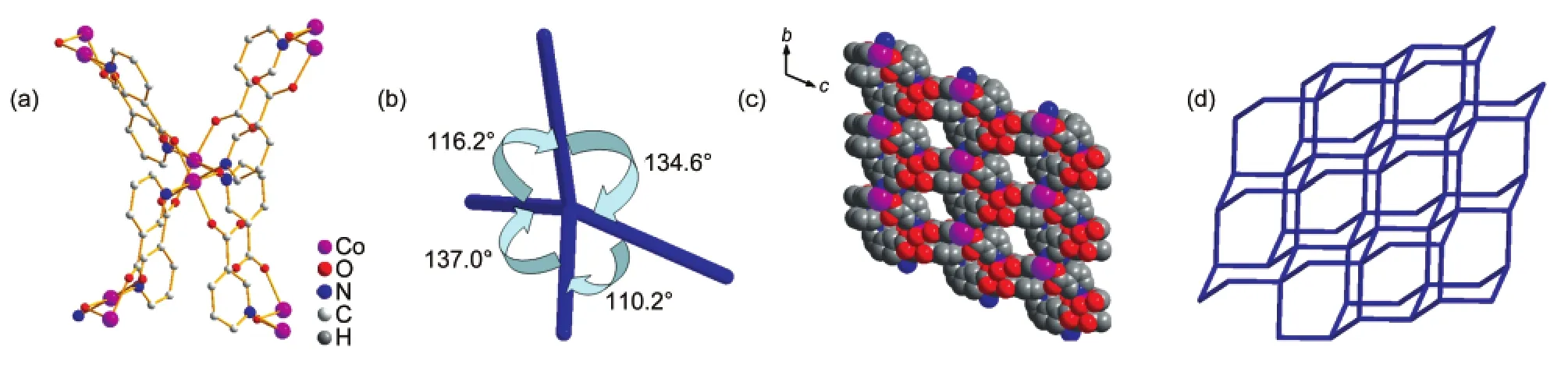
图2 化合物1的晶体结构Fig.2 Crystal structure of compound 1(a)the linkage of Co2(μ2-H2O);(b)the angles between the nodes consisted of Co2(μ2-H2O); (c)viewed along a axis,a 1D channel structure;(d)the topology of compound 1
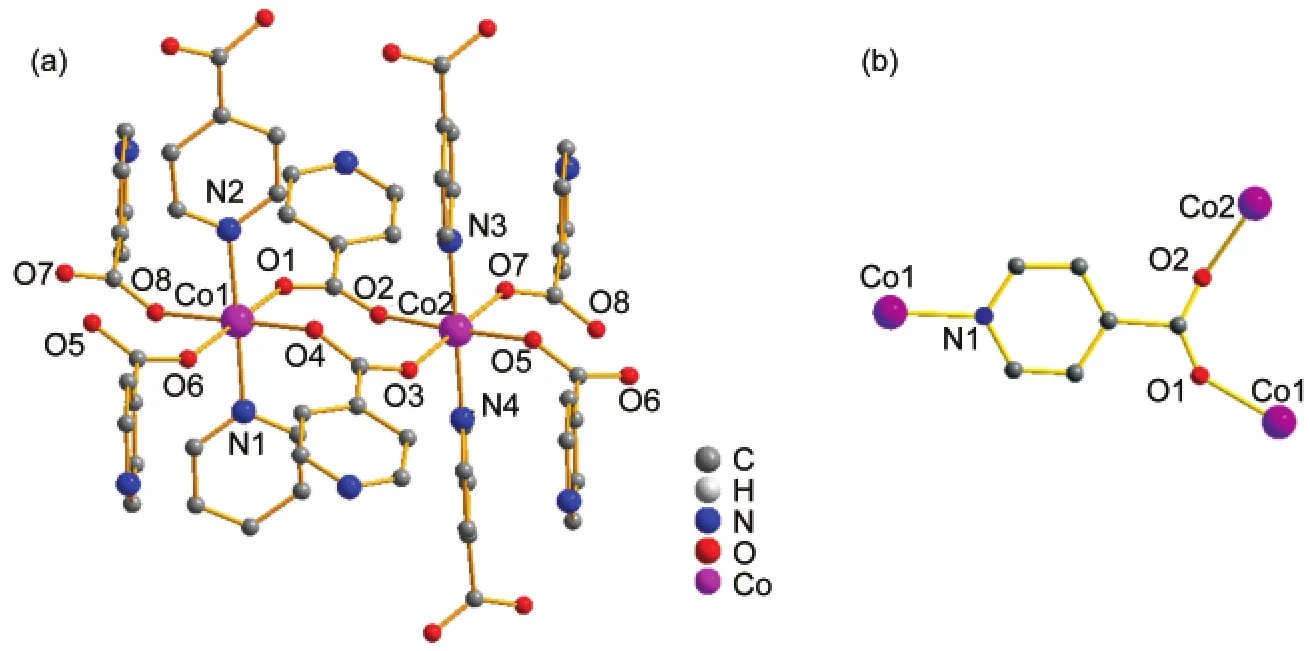
图3 化合物2中Co(II)的配位环境Fig.3 Coordination environment of Co(II)in compound 2(a)the coordination environment of Co1 and Co2;(b)the coordination mode of isonicotinic acid
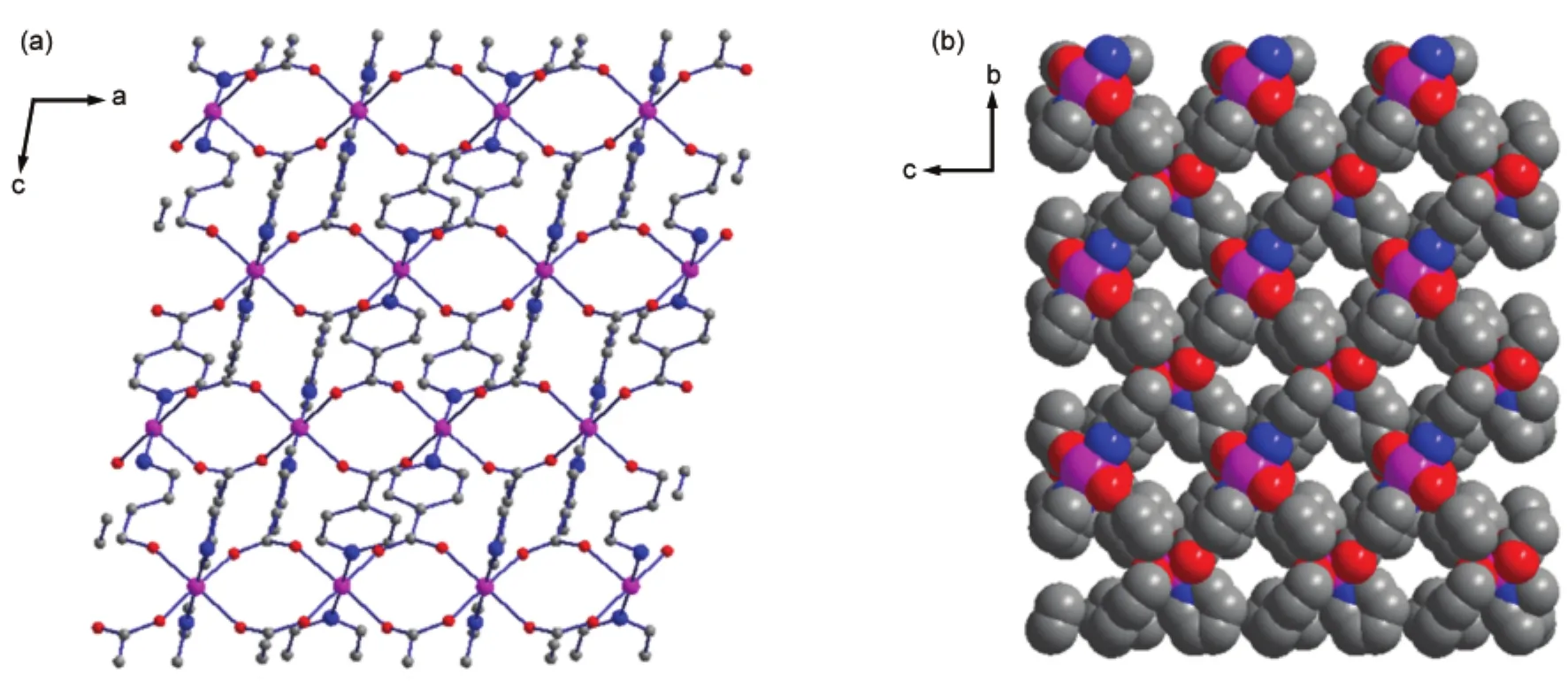
图4 化合物2的晶体结构Fig.4 Crystal structure of compound 2(a)chains along a axis;(b)packing diagram along bc plane,DMF molecules filled in 1D channels have been omitted for clarity.
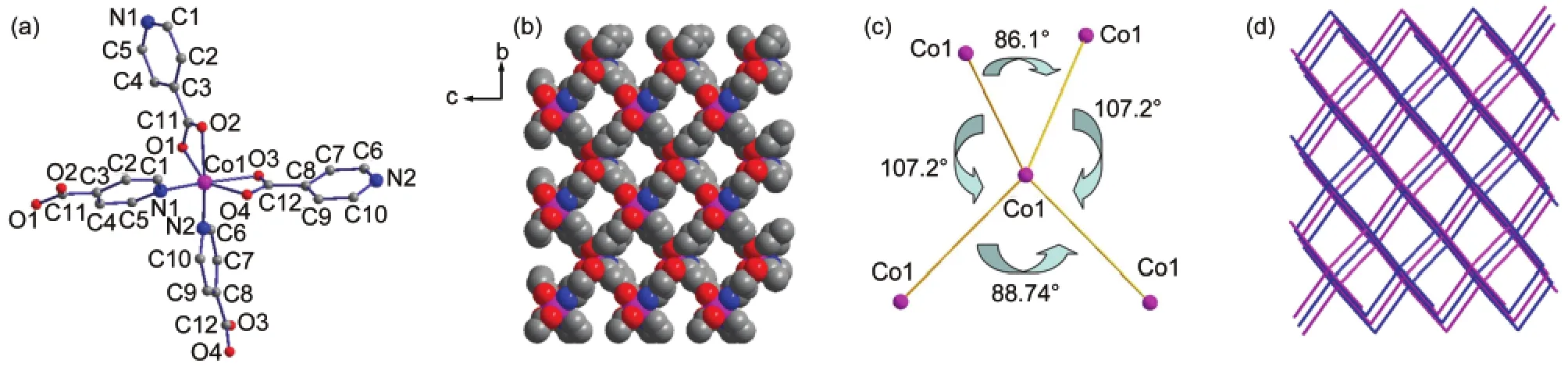
图5 化合物3的晶体结构Fig.5 Crystal structure of compound 3(a)coordination environment of Co1;(b)viewed along a axis,1D channel structure.DMF molecules filled in the channels have been omitted for clarity;(c)the angles between four-connected Co1;(d)topology of compound 3
将异烟酸与Co(NO3)2·6H2O在DMF中的反应温度升至160°C,则获得了与化合物2结构完全不同的化合物3,其晶体结构属于单斜晶系Pn空间群,主要键长见表2.该结构中只含有一种Co(II), Co1与两个羧酸根(l(Co1―O1)=0.2059 nm,l(Co1―O2)=0.2240 nm,l(Co1―O3)=0.2256 nm,l(Co1―O2)=0.2069 nm)及两个吡啶(l(Co1―N1)=0.2072 nm,l(Co1―N2)=0.2085 nm)配位(图5(a)).羧酸根的配位方式与化合物2中不同,两个羧酸根均与Co1形成双齿螯合配位.沿着a轴方向形成一维的孔道结构,孔道中填充的是DMF分子(图5(b)).整个结构中Co(II)与DMF分子的摩尔比为1:1.利用platon计算,溶剂DMF分子占据36.3%的空间.35将Co1视作四向连接的节点,其与周围四个Co1形成的角度为86.1°-107.2°(图5(c)),在空间中形成二重互穿的类金刚石型拓扑结构(图5(d)).
3.2 红外光谱
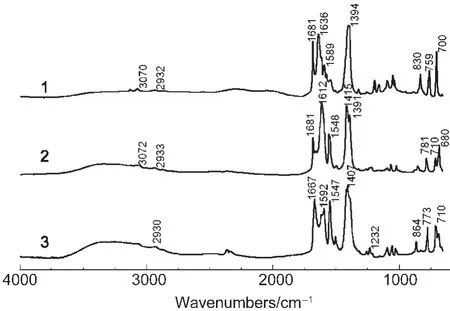
图6 化合物1-3的傅里叶变换红外(FTIR)光谱Fig.6 Fourier transform infrared(FTIR)spectra for compounds 1-3
图6为三种化合物的红外光谱.化合物1-3的红外谱图中均有DMF的C=O伸缩振动峰,其中化合物1和2中C=O的吸收峰均位于1681-1682 cm-1,而化合物3中C=O的吸收峰位于1667 cm-1,且相对于1400 cm-1附近羧酸根的对称伸缩振动峰的强度明显强于前两个.DMF吸收峰相对强度的不同,与处于孔道中DMF的相对比例有关,化合物3中DMF的含量最大,所以吸收峰相对强度最大.三种化合物羧酸根的反对称伸缩振动峰也明显不同:化合物1中位于1638 cm-1,且在1589 cm-1处存在较弱的肩峰;化合物2中该峰移动到了1612 cm-1;化合物3中存在1614和1592 cm-1两个吸收峰.羧酸根反对称伸缩振动峰的峰位不同,是由于三种化合物中羧酸根配位方式不同造成的.
3.3 热稳定性
图7中的热重曲线表明,化合物1在130-230°C区间失重14.5%,对应于1个DMF分子的失去,失去DMF的结构稳定到350°C开始分解.化合物2中DMF分子的失去从100°C开始持续到300°C,失重13.8%,该结构在400°C后彻底分解.化合物3中DMF的失去从室温开始持续到230°C,失重22.0%.
利用热重曲线的结果,将化合物1在200°C下真空处理6 h,对处理前后的样品分别测定了粉末X射线衍射数据和红外光谱.粉末X衍射数据表明200°C下处理后,化合物1的结构发生了一定程度的变化(图8).将加热处理后的化合物1浸入DMF两天后,结构又能回到未处理前的状态.上述过程在红外光谱(图9)中也能观察到,加热处理后,样品中位于1681 cm-1处DMF的C=O伸缩振动峰基本消失,而烟酸配体上羧酸根的反对称伸缩振动峰由1638 cm-1红移到1612 cm-1,表明DMF的脱附基本完成,羧酸根与Co(II)的配位发生一定的变化.而加热处理后的化合物1浸入DMF后其红外光谱与新制备的化合物1基本一致,表明化合物1脱附和吸附DMF的过程是可逆的.经过加热脱附处理的化合物1在77 K测定了N2的等温吸附-脱附曲线(图10),结果显示脱附处理后的化合物1在77 K,0.1 MPa下吸附N2的能力为55 cm3·g-1,计算出其BET比表面积为189 m2·g-1,其N2吸附-脱附曲线呈现典型的微孔结构的特征.
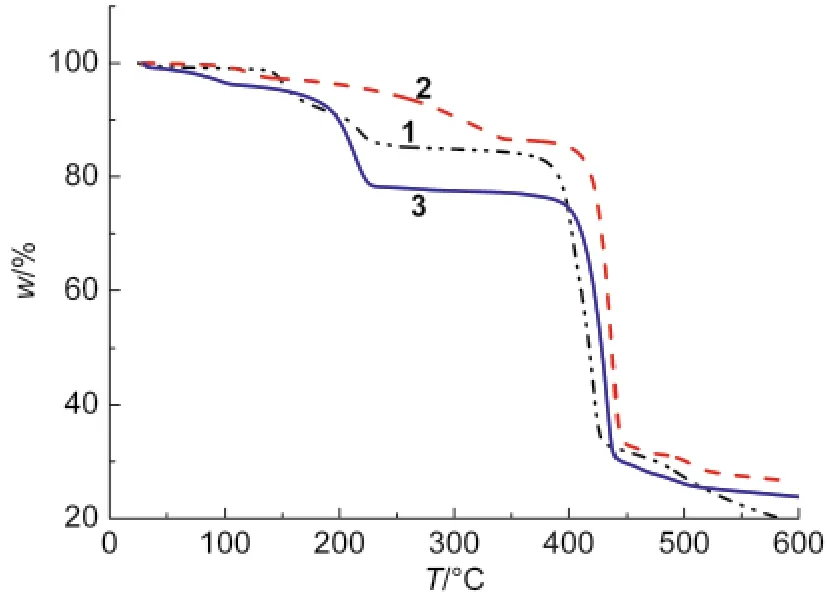
图7 化合物1-3的热重分析(TGA)曲线Fig.7 Thermogravimetric analysis(TGA)curves for compounds 1-3
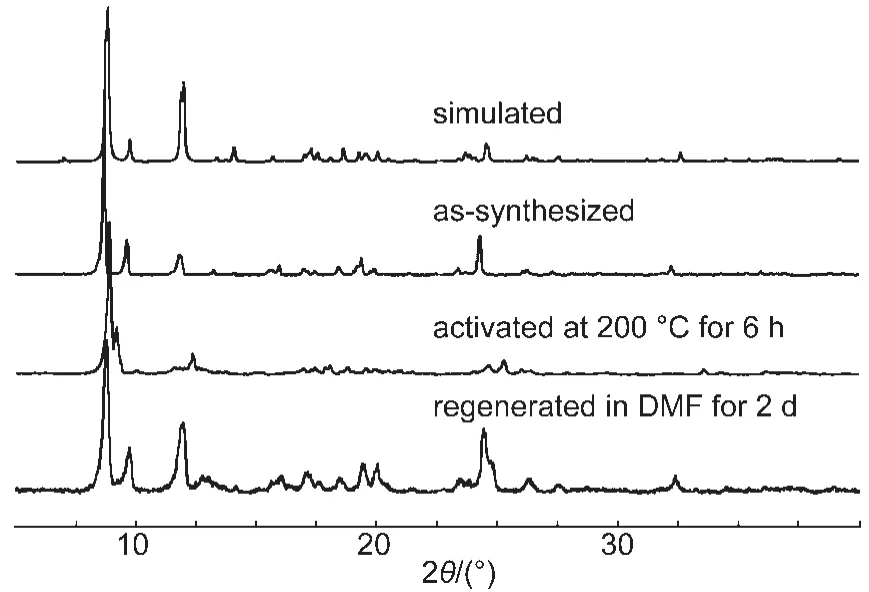
图8 化合物1的粉末X射线衍射(XRD)图Fig.8 Powder X-ray diffraction(XRD)patterns of compound 1
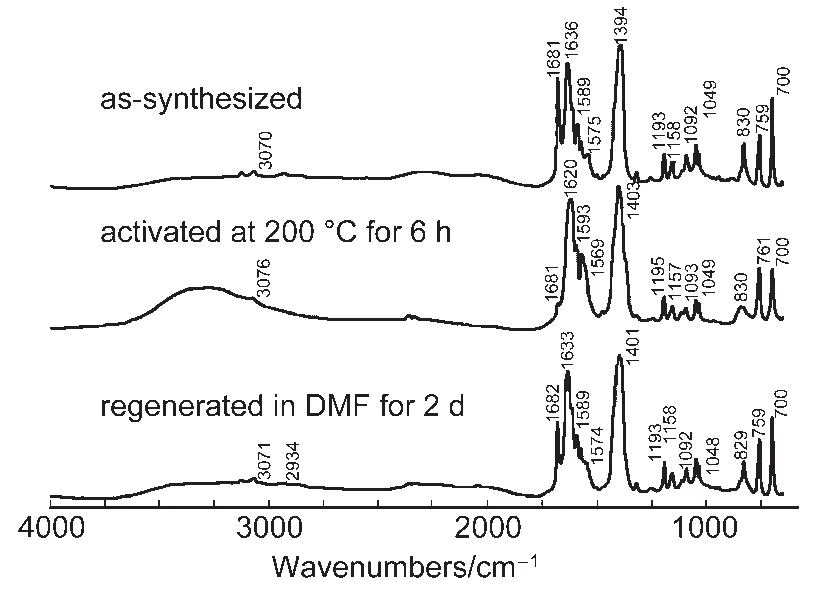
图9 化合物1的FTIR光谱图Fig.9 FTIR spectra for compound 1
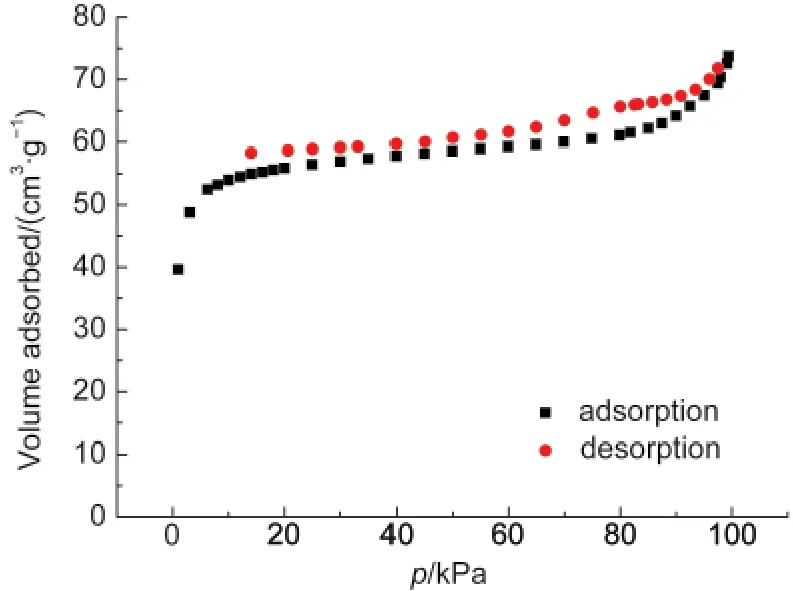
图10 化合物1在77 K时的N2吸附-脱附曲线Fig.10 N2adsorption-desorption curves forcompound 1 at 77 K
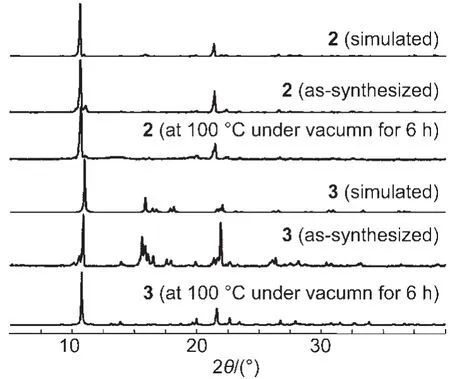
图11 化合物2和3的粉末XRD图Fig.11 Powder XRD patterns for compounds 2 and 3

图12 化合物2和3的FTIR光谱Fig.12 FTIR spectra for compounds 2 and 3
对化合物2和3进行了相似的处理.将上述两种化合物在100°C真空条件下加热6 h后,分别测定了粉末X射线衍射数据、红外光谱和N2吸附-脱附曲线.从图11的粉末X射线衍射数据可知,加热处理后化合物2的结构变化不大,而化合物3经加热处理后有向化合物2转变的趋势.图12的红外光谱显示加热处理后化合物2和3中DMF的C=O吸收峰基本消失.值得一提的是,经过加热,化合物3中羧酸根的反对称伸缩振动峰由1615和1592 cm-1两个吸收峰变成1612 cm-1的单峰,与化合物2中羧酸根的吸收峰位置基本相同.加热处理过程中化合物2和3的颜色均变成蓝紫色,结合红外光谱和粉末衍射数据,可以推测加热处理使得原本六配位的Co(II)有向四配位转变的趋势.对处理后的化合物2和3进行了N2吸附测试,计算出其BET比表面积分别为7.7和5.4 m2·g-1,结果表明,尽管化合物2和3晶体结构中的孔隙率较高,但在加热脱附DMF的过程中孔道结构均未能保持稳定.
4 结论
采用溶剂热法分别合成了烟酸和异烟酸与Co(II)的三种配位聚合物,对其单晶结构进行了分析,并测定了红外光谱和粉末X射线衍射以考察孔道的热稳定性.分析表明化合物1中由Co2(μ2-H2O)作为四向节点形成了类金刚石的三维结构.该结构中含有稳定的一维孔道,在200°C下脱附孔道中DMF分子后仍能稳定存在.化合物2和3是利用异烟酸与Co(II)分别在100°C和160°C两种不同温度下制得,在两种化合物中配体和Co(II)采用完全不同的配位模式,导致其具有不同的三维结构.但化合物2和3中的一维孔道在加热脱附DMF分子的过程中均不能稳定存在,化合物3在加热处理中有向化合物2转变的趋势.
(1) Kitagawa,S.;Uemura,K.Chem.Soc.Rev.2005,34,109.doi: 10.1039/b313997m
(2) Kitagawa,S.;Matsuda,R.Coord.Chem.Rev.2007,251,2490. doi:10.1016/j.ccr.2007.07.009
(3) James,S.L.Chem.Soc.Rev.2003,32,276.doi:10.1039/ b200393g
(4)Tranchemontagne,D.J.;Mendoza-Cortes,J.L.;OʹKeeffe,M.; Yaghi,O.M.Chem.Soc.Rev.2009,38,1257.doi:10.1039/ b817735j
(5) Chen,B.L.;Yang,Y.;Zapata,F.;Lin,G.N.;Qian,G.D.; Lobkovsky,E.B.Adv.Mater.2007,19,1693.doi:10.1002/ adma.200601838
(6) Chen,B.;Wang,L.;Xiao,Y.;Fronczek,F.R.;Xue,M.;Cui,Y.; Qian,G.Angew.Chem.Int.Edit.2009,48,500.doi:10.1002/ anie.200805101
(7) Rocha,J.;Carlos,L.D.;Paz,F.A.A.;Ananias,D.Chem.Soc. Rev.2011,40,926.doi:10.1039/c0cs00130a
(8) Liu,T.F.;Fu,D.;Gao,S.;Zhang,Y.Z.;Sun,H.L.;Su,G.;Liu, Y.J.J.Am.Chem.Soc.2003,125,13976.doi:10.1021/ ja0380751
(9)Zhang,B.;Wang,Z.M.;Kurmoo,M.;Gao,S.;Inoue,K.; Kobayashi,H.Adv.Funct.Mater.2007,17,577.doi:10.1002/ adfm.200600265
(10) Salles,F.;Maurin,G.;Serre,C.;Llewellyn,P.L.;Knofel,C.; Choi,H.J.;Filinchuk,Y.;Oliviero,L.;Vimont,A.;Long,J.R.; Ferey,G.J.Am.Chem.Soc.2010,132,13782.doi:10.1021/ ja104357r
(11) Guillou,N.;Livage,C.;Drillon,M.;Ferey,G.Angew.Chem. Int.Edit.2003,42,5314.doi:10.1002/anie.200352520
(12) Hu,A.;Ngo,H.L.;Lin,W.B.Angew.Chem.Int.Edit.2003,42, 6000.doi:10.1002/anie.200351264
(13)Kesanli,B.;Lin,W.B.Chem.Commun.2004,2284.
(14) Horcajada,P.;Surble,S.;Serre,C.;Hong,D.Y.;Seo,Y.K.; Chang,J.S.;Greneche,J.M.;Margiolaki,I.;Ferey,G.Chem. Commun.2007,2820.
(15) Kitagawa,S.;Kitaura,R.;Noro,S.Angew.Chem.Int.Edit. 2004,43,2334.doi:10.1002/anie.200300610
(16) Rosi,N.L.;Eckert,J.;Eddaoudi,M.;Vodak,D.T.;Kim,J.; OʹKeeffe,M.;Yaghi,O.M.Science 2003,300,1127.doi: 10.1126/science.1083440
(17)Li,H.;Eddaoudi,M.;OʹKeeffe,M.;Yaghi,O.M.Nature 1999, 402,276.doi:10.1038/46248
(18) Ferey,G.Chem.Soc.Rev.2008,37,191.doi:10.1039/b618320b
(19)Suh,M.P.;Park,H.J.;Prasad,T.K.;Lim,D.W.Chem.Rev. 2011,112,782.
(20) Li,J.R.;Sculley,J.;Zhou,H.C.Chem.Rev.2012,112,869. doi:10.1021/cr200190s
(21)Sumida,K.;Rogow,D.L.;Mason,J.A.;McDonald,T.M.; Bloch,E.D.;Herm,Z.R.;Bae,T.H.;Long,J.R.Chem.Rev. 2012,112,724.doi:10.1021/cr2003272
(22) Fu,Y.;Li,G.B.;Liao,F.H.;Lin,J.H.Acta Phys.-Chim.Sin. 2011,27,2269.[付 颖,李国宝,廖复辉,林建华,物理化学学报,2011,27,2269]doi:10.3866/PKU.WHXB20110908
(23)Yaghi,O.M.;Davis,C.E.;Li,G.;Li,H.J.Am.Chem.Soc. 1997,119,2861.doi:10.1021/ja9639473
(24)Rosi,N.L.;Kim,J.;Eddaoudi,M.;Chen,B.L.;OʹKeeffe,M.; Yaghi,O.M.J.Am.Chem.Soc.2005,127,1504.doi:10.1021/ ja045123o
(25) Fujita,M.;Kwon,Y.J.;Washizu,S.;Ogura,K.J.Am.Chem. Soc.1994,116,1151.doi:10.1021/ja00082a055
(26) Kepert,C.J.;Rosseinsky,M.J.Chem.Commun.1999,375.
(27) Noro,S.;Kitaura,R.;Kondo,M.;Kitagawa,S.;Ishii,T.; Matsuzaka,H.;Yamashita,M.J.Am.Chem.Soc.2002,124, 2568.doi:10.1021/ja0113192
(28) Li,Y.;Xie,L.;Liu,Y.;Yang,R.;Li,X.Inorg.Chem.2008,47, 10372.doi:10.1021/ic800919k
(29) Park,H.J.;Suh,M.P.Chem.Commun.2010,46,610.doi: 10.1039/b913067e
(30)Chun,H.;Dybtsev,D.N.;Kim,H.;Kim,K.Chem.Eur.J.2005, 11,3521.doi:10.1002/chem.200401201
(31) Xiang,S.;Huang,J.;Li,L.;Zhang,J.;Jiang,L.;Kuang,X.;Su, C.Y.Inorg.Chem.2011,50,1734.
(32) Eubank,J.F.;Wojtas,L.;Hight,M.R.;Bousquet,T.;Kravtsov, V.C.;Eddaoudi,M.J.Am.Chem.Soc.2011,133,17532.doi: 10.1021/ja203898s
(33)Sheldrick,G.M.SHELXS-97,A Computer Program for the Solution of Crystal Structures;University of Göttingen: Göttingen,Germany,1997.
(34)Sheldrick,G.M.SHELXL-97,A Computer Program for Refinement of Crystal Structures;University of Göttingen: Götingen,Germany,1997.
(35) Spek,A.L.PLATON,A Multipurpose Crystallographic Tool; Utrecht University:Utrecht,The Netherlands,2001.
(36)Ayyappan,P.;Evans,O.R.;Lin,W.B.Inorg.Chem.2001,40, 4627.
(37) Liu,Y.H.;Tsai,H.L.;Lu,Y.L.;Wen,Y.S.;Wang,J.C.;Li,K. L.Inorg.Chem.2001,40,6426.doi:10.1021/ic010343d
May 18,2012;Revised:June 12,2012;Published on Web:June 12,2012.
Crystal Structures and Pore Stability of Coordination Polymers Constructed by Nicotinic Acid and Isonicotinic Acid with Co(II)
HUA Xiao-Hui LI Wei-Hong*XU Yi-Zhuang WU Jin-Guang
(College of Chemistry and Molecular Engineering,Peking University,Beijng 100871,P.R.China)
Nicotinic acid and isonicotinic acid were utilized to reactwith Co(NO3)2in N,Nʹ-dimethylformamide(DMF)in solvothermal conditions respectively,and resulted in three new coordination polymers:[Co2(μ2-H2O)(nicotinic acid)4·(DMF)](1),[Co2(isonicotinic acid)4·(DMF)](2),and[Co(isonicotinic acid)2·(DMF)](3).Single crystal X-ray diffraction(XRD)and elemental analysis were performed to obtain structuralinformation on these compounds.Fourier transform infrared (FTIR) spectroscopy, thermogravimetric analysis(TGA),and N2isotherm analysis were utilized to investigate the pore stability of the three compounds.The above experiments reveal that compound 1 has a diamond-like topology,and the one-dimensional(1D)channels in compound 1 are stable after the removal of DMF molecules. Compounds 2 and 3 were synthesized at 100 and 160°C by the coordination of isonicotinic acid with Co(II),respectively.The coordination modes of isonicotinic acid to Co(II)are completely different in the two compounds,which lead to two different crystal structures.The 1D channels in compounds 2 and 3 were not stable after the removal of DMF molecules.
Coordination polymer;Nicotinic acid;Crystal structure;Topology;Fourier transform infrared spectroscopy; Pore stability
10.3866/PKU.WHXB201206123
∗Corresponding author.Email:Weihong.Li@pku.edu.cn;Tel:+86-10-62751238.
The project was supported by the National Natural Science Foundation of China(20671007).
国家自然科学基金(20671007)资助项目
O641
