B12Sc4和B12Ti4团簇的储氢性质
2012-12-21马丽娟王剑锋贾建峰武海顺
马丽娟 王剑锋 贾建峰 武海顺
(山西师范大学化学与材料科学学院,山西临汾041004)
B12Sc4和B12Ti4团簇的储氢性质
马丽娟 王剑锋 贾建峰*武海顺
(山西师范大学化学与材料科学学院,山西临汾041004)
提出了两个稳定的团簇B12Sc4和B12Ti4,基于理论计算,研究了它们的结构与储氢性质.结果发现,在这两个稳定的团簇中,过渡金属原子不会聚合在一起而影响它们对氢气的吸附.B12Sc4最多可以吸附12个氢分子,达到7.25%(质量分数)的储氢量,它的平均每氢分子吸附能量为-10.5 kJ·mol-1.B12Ti4最多只能吸附8个氢分子,储氢量为4.78%,但其平均每氢分子吸附能量可达-50.2 kJ·mol-1.进一步计算表明,即使在77 K,也需要很高的氢气压力才能使12个氢分子都吸附到B12Sc4上.电子结构分析表明,B12Ti4-nH2吸附结构中的Kubas作用要大于相应B12Sc4-nH2结构中的Kubas作用.
硼团簇;金属掺杂;储氢;吸附;从头算
1 Introduction
Hydrogen is widely viewed as the next generation of energy carrier to replace the fossil fuels due to its abundance,high chemical energy,and pollution-free burning.1-3However,hydrogen storage is a“bottleneck”for the on-board application of hydrogen as energy carrier.Several ways have been investigated and developed to store hydrogen gas,involving its compression,liquefaction,and adsorption in several metals and metal alloys and so on.Unfortunately,none of these technologies are good enough to satisfy the on-board application of hydrogen energy,even though each way possesses desirable characteristics in certain areas.4-7For example,very high pressure vessels are capable of storing hydrogen about 9%(mass fraction),however,so high pressure will bring serious security problem.The hydrogen storage capacities of many complex hydrides,such as Li3Be2H7,8NaAlH49and so on,10-12in which hydrogen atoms are chemical bonded to metals,are beyond about 6%.However they can not be easily,quickly recovered when exhausted.Highly porous carbon materials13-15and metal organic frameworks(MOF)16,17represent another type of hydrogen storage material,which interact with hydrogen physically,and are kineticly favorable for recharge of hydrogen.However, they can achieve high storage capacity only under very low temperature.At ambient temperature,these materials barely adsorb hydrogen due to weak interaction between hydrogen and the solid materials.
Zhao et al.18proposed that the fullerene decorated by transition metal may be a good candidate for hydrogen storage. Based on theoretical calculations,they found that Sc decorated C60and C48B12are capable of storing hydrogen about 7%and 8.77%at ambient condition.At the same time,Yildirim and Ciraci19showed that Ti decorated carbon nanotube can approach about 8%hydrogen storage capacity.In both Sc decorated C60and Ti decorated carbon nanotube,the transition metal atoms scatter on the surface of carbon nanomaterials,and bind with hydrogen molecules through Kubas interaction.20
Unfortunately,Sunʹs theoretical investigation21indicated that transition metal atoms on C60will be clustered rather than scattered,and the clustering of the transition metal atoms will significantly reduce the mass percentage of hydrogen storage.Sun et al.22,23found that the clustering problem can be resolved by replacing the transition metal atoms with alkali metal atoms or alkaline-earth metal atoms.However,alkali or alkaline-earth metal doped fullerene can only be used in low temperature due to the small binding energy of hydrogen molecule to them.24,25Moreover,the recent theoretical study shows some alkali metal atoms,such as Na,also will be aggregated on the surface of C60.26
Meng et al.27found that the Ti atoms on the boron nanotube, contrary to on carbon nanostructure,will keep in isolation from each other.Theoretical calculations showed that metal doped icosahedral B80also does not suffer the clustering problem,being a promising hydrogen storage candidate.28-31
However,more recent theoretical calculations indicated that the most stable B80and other medium-sized boron clusters have core-shell rather than hollow cage structure.32Zhao and his coworkers33have designed a new type of hydrogen storage media,chained TiBx.They found that the most stable TiB5chain can reach 7.3%hydrogen storage capacity with the average binding energy per H2of 43.7 kJ·mol-1.
In this work,we propose a Sc doped B12cluster and a Ti doped B12cluster,inspired by our previous foundation of a B12core in B12CO12.34The structures of B12Sc4and B12Ti4,as well as their interactions with H2are discussed in detail.
2 Computational methods
All the isomers were optimized at the level of density functional theory(DFT)with Beckeʹs three-parameter exchange35and Lee-Yang-Parr correlation functional36by using Gaussian 03 program.37The standard split valence basis set 6-31G(d,p) was employed to describe the orbitals of all atoms involved. Geometry optimizations were done with no symmetry restriction.All the reported isomers were characterized at the same level as energy minima by frequency calculations.All the population analyses were based on the data obtained at the B3LYP/ 6-31G(d,p)level.Many investigations demonstrated that MP2 method was more reliable for calculating the weak interaction.38,39So,to obtain accurate average binding energy of H2to B12Sc4or B12Ti4,the single point energy calculations for all B12X4-nH2(X=Sc,Ti),the most stable B12X4and H2were performed at the MP2/6-311G(d,p)level.The average binding energy per H2(ABE/H2)was defined as
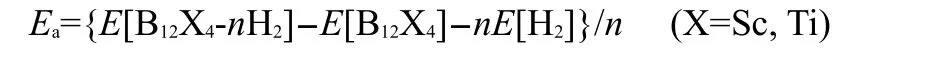
where,E[B12X4],E[H2],and E[B12X4-nH2]are the electronic energies of relaxed B12X4,H2,and B12X4-nH2,respectively;and n is the number of H2molecules.The basis set superposition error(BSSE)has been corrected using the full counterpoise method for all the B12X4-nH2complexes at the MP2/6-311G(d, p)level.
3 Results and discussion
3.1 Structures of B12Sc4and B12Ti4
Our proposed B12Sc4cluster is shown as isomer 2 in Fig.1, which has D2dsymmetry,and was optimized from an initio structure,isomer 1.Isomer 1 is originated from a stable(BCO)12boron carbonyl compound.29The multiplicity of isomer 2 is 1.To confirm that it is the favorable structure for B12Sc4,other B12Sc4isomers are considered in our work.Isomer 3 in Fig.1 is constructed from the most stable B12and Sc4clusters.The geometry optimization of isomer 3 gives out the isomer 4,which is less stable than 2 about 703.4 kJ·mol-1at the B3LYP/6-31G(d, p)level.Based on the most stable quasiplanar B12and icosahedral B12,40we put four Sc atoms on it in all possible patterns,all these structures were optimized to amorphous structure with higher energy than isomer 2(see Fig.S1 in Supporting Information for their structures and energies).In isomer 2,it is clear that the Sc atoms do not cluster together.
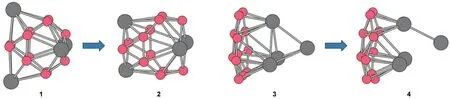
Fig.1 Initio and optimized structures of two B12Sc4isomers
Ab initio molecular dynamic(MD)simulation also confirmed that our proposed B12Sc4is stable.The isomer 2 was simulated using 0.5 fs time step in a 14×14×14 simple cubic supercell with Born-Oppenheimer MD implemented in CP2k code.41First,the system was equilibrated at 1000 K in a NVE ensemble with a temperature tolerance of 500 K within 10000 steps.When it achieved equilibrium,we continued the MD at the same condition for another 5000 steps(see Supporting Information for more detail information about the MD calculation configurations).The result shows that our proposed B12Sc4is intact throughout the whole simulation.A similar MD simulation shows that the IhB80Sc12will collapse to a core-shell structure.We expect that our proposed B12Sc4is stable enough at their possible operation temperatures about 300 K.
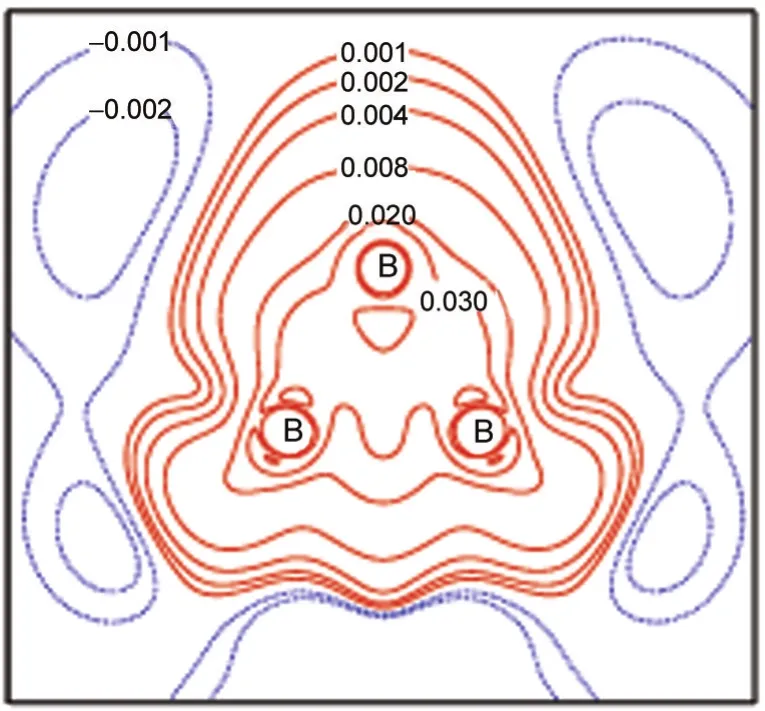
Fig.2 Electronic difference density(in×103nm-3)map of a B3ring in B12Sc4
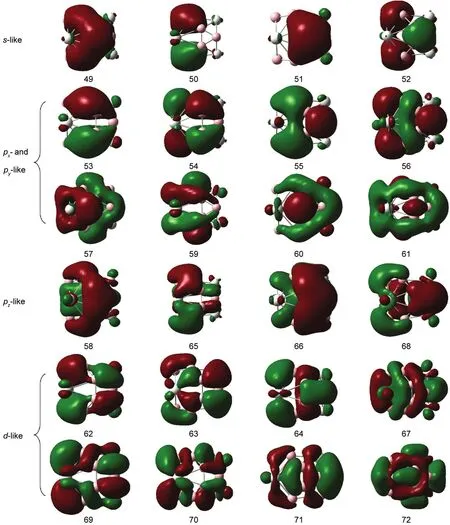
Fig.3 Occupied valance orbitals of B12Sc4
To understand the stability of B12Sc4more foundationally,we has performed a detail inspect about the structure of B12Sc4. The energy gap between the highest occupied molecular orbital and the lowest unoccupied molecular orbital(HOMO-LUMO) of B12Sc4is 2.17 eV.In B12Sc4,12 boron atoms form four three-membered rings(B3).The electronic density difference map(Fig.2)of B3ring shows that electrons mainly shift to the center of the ring.It is clear that an open three-center bond is formed other than a strained‘banana bond’.So,B3rings are non-strained rings in B12Sc4.
Fig.3 lists all the valance orbitals of B12Sc4,which clearly shows that every B3ring,can be considered as a unit,possesses s-like,p-like,and d-like orbitals,when interacting with Sc atoms.Orbitals 49-52 of B12Sc4are formed by the s-like orbitals of B3units,without any node among the electronic cloud within B3rings.Orbitals 53-61,65,66,and 68 are originated from the p-like orbitals,with only one node among the electronic cloud within every B3ring.Moreover,the p-like orbitals of B3can be classified into px-,py-,and pz-like orbitals.In pz-like orbitals(58,65,66,68),the nodal plane is substantially parallel to the B3ring,while in px-and py-like orbitals,the nodal plane is nearly vertical to the B3ring.It should be noticed that some d-like components hybridize into the orbitals 65 and 66 when it interacts with Sc atoms.Orbitals 62-64,67,and 69-72 are mainly formed by the d-like orbital of B3units with some p-like components hybridizing into orbitals 62-64 significantly.
The B12Ti4has the same topological structure as isomer 2, and has D2dsymmetry and no unpaired electron.The other isomers constructed based on the most stable quasiplanar B12and icosahedral B12as for B12Sc4are also considered(see Fig.S2 in Supporting Information).They are all higher in energy than isomer 2.The same MD simulation as for B12Sc4was performed for it,which also demonstrates that B12Ti4are stable around 1000 K.The most obvious distinction between B12Sc4and B12Ti4is their metal-metal(M-M)distances.The adjacent Sc-Sc distance in B12Sc4is 0.315 nm while Ti-Ti distance in B12Ti4is only 0.294 nm.Too short M-M distance is unfavorable for hydrogen storage.The HOMO-LUMO energy gap of B12Ti4is only 1.21 eV,nearly the half of B12Sc4.
3.2 Hydrogen molecule adsorption on B12Sc4and
B12Ti4
We now turn to the discussion on the adsorption of hydrogen molecules on the B12Sc4and B12Ti4clusters mentioned above. The optimized adsorbing structures with 1,2,and 3 hydrogen molecules on each Sc atom of B12Sc4are shown in Fig.4 as isomer 5(B12Sc4-4H2),6(B12Sc4-8H2),7(B12Sc4-12H2).The adsorbing structure of B12Ti4with one H2on each Ti atom(B12Ti4-4H2) is the same as the corresponding B12Sc4-4H2.B12Ti4can adsorb 8 hydrogen molecules at most.The optimized B12Ti4-8H2is presented in Fig.4 as isomer 8,which is slightly different from B12Sc4-8H2.The average binding energies per hydrogen molecule(ABE/H2),the largest and shortest Sc-H(Ti-H)distances,the bond lengths of hydrogen molecules for both B12Sc4-nH2and B12Ti4-nH2(n=4,8,12 for B12Sc4;n=4,8 for B12Ti4)are listed in Table 1.
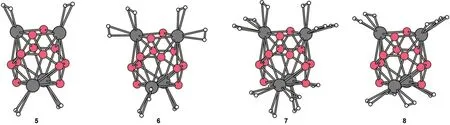
Fig.4 Structures of B12Sc4-nH2and B12Ti4-nH25:B12Sc4-4H2,6:B12Sc4-8H2,7:B12Sc4-12H2,8:B12Ti4-8H2
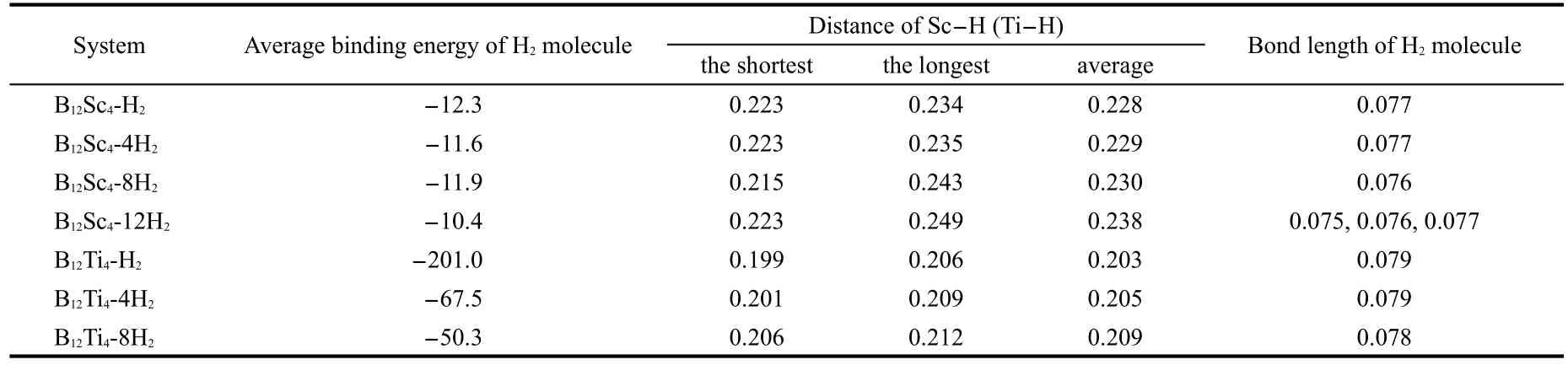
Table 1 Average binding energies of H2molecule(in kJ·mol-1per H2),M-H distances(M=Sc,Ti),and bond lengths of H2in B12Sc4-nH2and B12Ti4-nH2(in nm)
Now,we begin to discuss the interaction between B12Sc4and H2molecules.In B12Sc4-4H2(5),the shortest Sc-H distance is 0.223 nm,and the average Sc-H distance is 0.229 nm.These Sc-H distances are longer than the normal Sc―H bond length (about 0.18 nm in ScH2molecule42,43).The ABE/H2of B12Sc4-4H2(5)is only-11.6 kJ·mol-1,whereas we have noted that in B80Sc-H2,the binding energy of H2to B80Sc is-42.5 kJ·mol-1.25The large difference of ABE/H2between B12Sc4-4H2and B80Sc-H2indicates that the structure of the BxScydoped cluster has a significant effect on the binding energy of H2adsorbed on it.In B12Sc4-4H2(5),the H―H bond length is 0.077 nm,which is longer than the bond length of an isolated hydrogen molecule (0.074 nm optimized at the B3LYP/6-31G(d,p)level).On C60Sc12,the first H2will dissociate when binding to a Sc atom.13The elongation of the H-H bond length is caused both by the Kubas interaction and induced interaction.We have calculated the K+-H2and Ca2+-H2systems at the B3LYP/6-31G(d,p)level to estimate the effect of the induced interaction to the H-H bond.The result shows that the H-H bond lengths of K+-H2and Ca2+-H2are 0.075 and 0.076 nm,respectively,which both are slightly longer than 0.074 nm of a free H2molecule,and smaller than that of B12Sc4-4H2.The ABE/H2of B12Sc4-8H2(6) and B12Sc4-12H2(7)are-11.9 and-10.4 kJ·mol-1,respectively. With three hydrogen atoms on each Sc atom,the hydrogen storage capacity of B12Sc4-12H2can reach to 7.25%.
As regards B12Ti4,it can accommodate for 8 hydrogen molecules at most,and reach to 4.78%hydrogen storage capacity only.However,the ABE/H2of B12Ti4-nH2is significantly greater than that of corresponding B12Sc4-nH2,as shown in Table 1. In B12Ti4-4H2,the Ti-H distances range from 0.201 to 0.209 nm,and have an average value of 0.205 nm,being longer than the normal Ti―H bond.The calculated Ti―H bond length in TiH4is about 0.170 nm,44and the experimental values range from 0.169 to 0.184 nm in different compounds.45,46The H-H distance of B12Ti4-4H2is 0.079 nm,which is 0.005 nm longer than thatoffree hydrogen molecule. The ABE/H2of B12Ti4-4H2is-67.5 kJ·mol-1,which is remarkably larger than that of B12Sc4-4H2.The large ABE/H2of B12Ti4-4H2may raise a doubt that the hydrogen molecule will dissociate to atoms in B12Ti4-H2.So,additional calculation about B12Ti4-H2is performed.As a comparison,B12Sc4-H2is also considered.Natural bond orbital(NBO)analysis indicates that the H―H bond is hold in B12Ti4-H2,with a bond length of 0.079 nm.However, the ABE/H2of B12Ti4-H2reaches to-201.0 kJ·mol-1,making it difficult to release the H2molecule.Considered that B12Ti4can adsorb 8 hydrogen molecules at most,it can conclude that B12Ti4is not a good candidate for hydrogen storage.The ABE/ H2of B12Sc4-H2is-12.3 kJ·mol-1,which being similar to that of other B12Sc4-nH2.
A detail analysis about the orbitals of B12Sc4-4H2and B12Ti4-4H2can account for why the ABE/H2of B12Ti4-nH2is greater than that of B12Sc4-nH2.Given that the charge of Ti atoms (NBO charge:1.26|e|)in B12Ti4is smaller than that of Sc atoms (NBO charge:0.95|e|)in B12Sc4,It is rational to conclude that the Kubas interaction in B12Ti4-nH2is more strong than that in B12Sc4-nH2.In B12Sc4-nH2and B12Ti4-nH2,the Kubas interaction involves the interaction of the σ-bonds of H2molecules to unoccupied orbitals of Sc and Ti atoms,as well as the σ*-bonds of H2molecules to occupied orbitals of Sc and Ti atoms.
In Fig.5,we give out the orbitals mainly located on hydrogen molecules both of B12Sc4-4H2and B12Ti4-4H2.For B12Sc4-4H4,the contributions from hydrogen molecules to these orbitals are in a range of 87%to 91%.The contributions from Sc atoms are all below 10%.However,for B12Ti4-4H4,the contributions from hydrogen molecules to these orbitals are in a range of 78%to 85%.The contributions from Ti atoms are in a range of 12%to 15%.The components of these orbitals clearly indicate that there are more electrons in σ-bonds of H2molecules transferred to the d orbital of metal atoms in B12Ti4-4H4than that in B12Sc4-4H4.
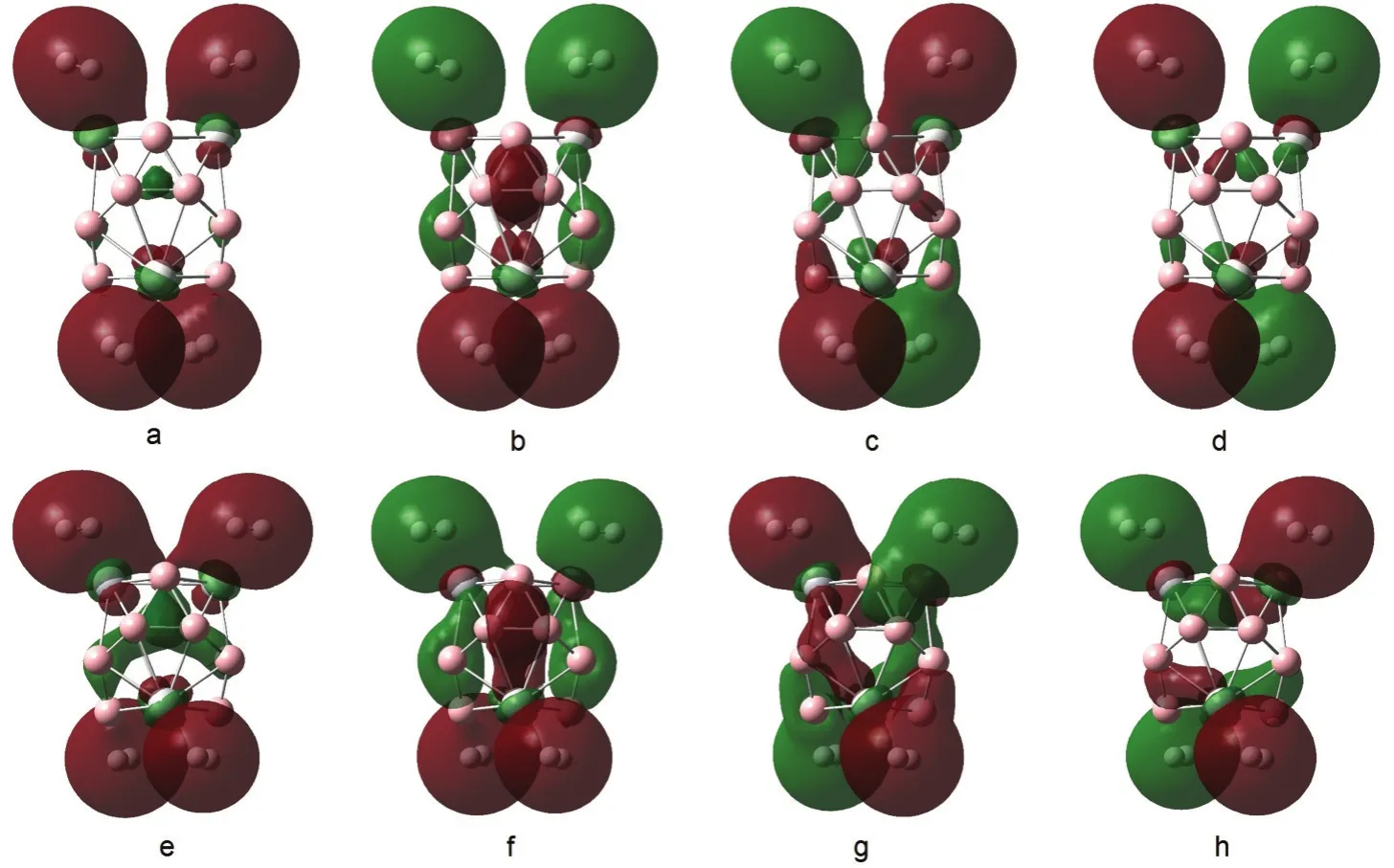
Fig.5 Orbitals mainly located on hydrogen molecules in B12Sc4-4H2and B12Ti4-4H2 for B12Sc4-4H2,a:HOMO-22,b:HOMO-21,c:HOMO-20,d:HOMO-19;for B12Ti4-4H2,e:HOMO-24,f:HOMO-23,g:HOMO-22,h:HOMO-21
Nearby the HOMO orbital,there are five orbitals involving the σ*-bonds of H2molecules and more than 5%contributions from hydrogen molecules are found for B12Ti4-4H4.They are HOMO,HOMO-1,HOMO-2,HOMO-4,and HOMO-5,as listed in Fig.6.The components from H2molecules in these five orbitals are 5.6%,14.0%,5.2%,9.0%,and 9.2%,respectively.For B12Sc4-4H4,only two similar types of orbitals are found.They are HOMO-1 and HOMO-2.The components from H2molecules in these two orbitals are 6.4%and 8.6%,respectively.The components of these orbitals indicate that there are more electrons from the d orbital of metal atoms donated back to the σ*-bonds of H2molecules in B12Ti4-4H4than that in B12Sc4-4H4.
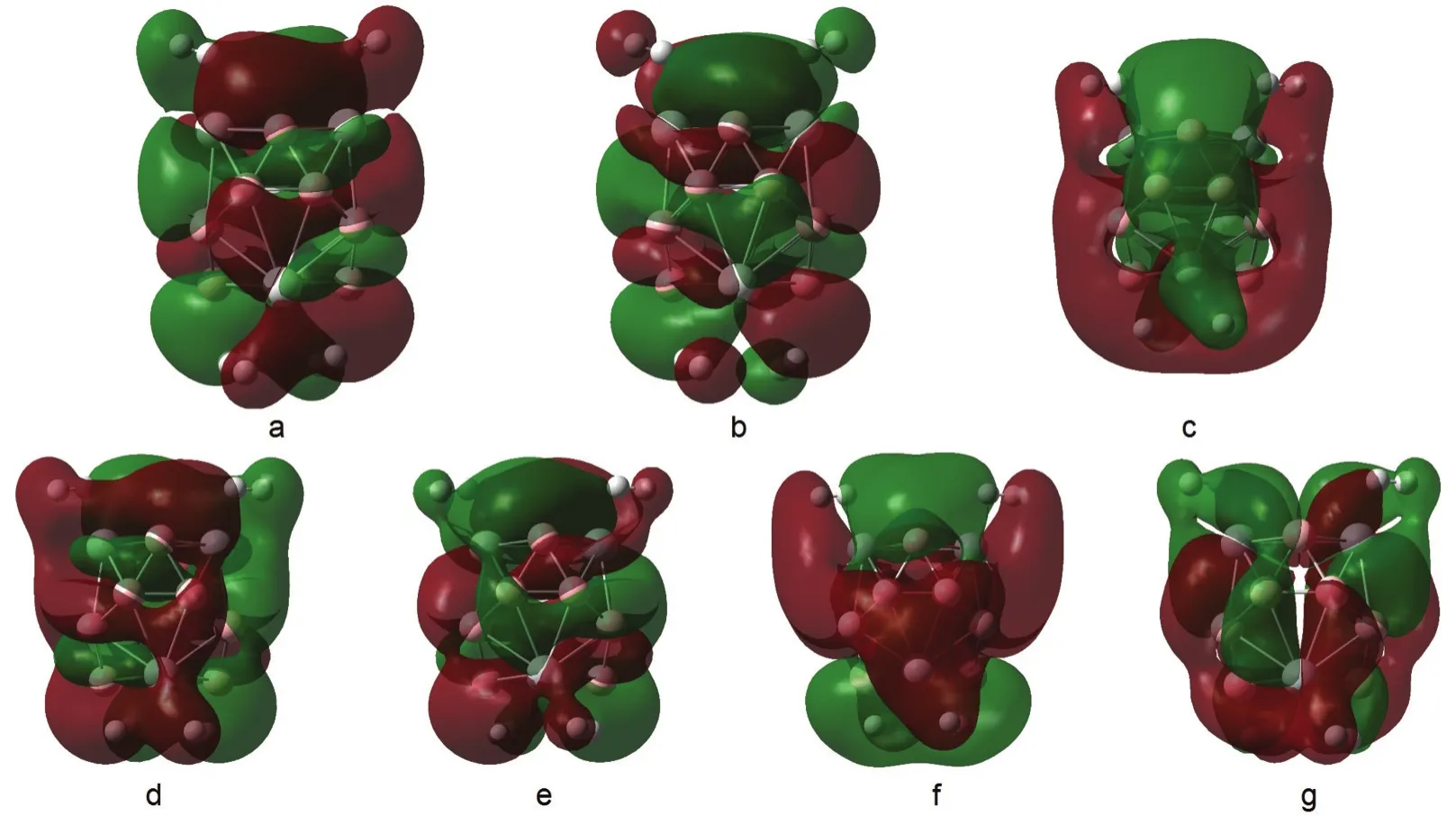
Fig.6 Orbitals involving the σ*-bonds of H2molecules in B12Sc4-4H2and B12Ti4-4H2for B12Sc4-4H2,a:HOMO-2,b:HOMO-1;for B12Ti4-4H2,c:HOMO-5,d:HOMO-4,e:HOMO-2,f:HOMO-1,g:HOMO
Above discussion clearly indicates that the Kubas interaction in B12Ti4-4H2is much stronger than that in B12Sc4-4H2.So, it is not surprising that the ABE/H2of B12Ti4-nH2is greater than that of B12Sc4-nH2due to the stronger Kubas interaction.The strong Kubas interaction in B12Ti4-nH2also is implied by the longer H―H bond length.As listed in Table 1,the H―H bond lengths are 0.079 and 0.078 nm in B12Ti4-4H2and B12Ti4-8H2, respectively,being longer than those in B12Sc4-4H2(0.077 nm) and B12Sc4-8H2(0.076 nm).
Now,it is necessary to consider the influences of the temperature and the pressure of hydrogen on the process of the hydrogen storage and release.The changes of Gibbs free energy (ΔG)for the process of B12Sc4+12H2→B12Sc4-12H2were calculated at 77 and 300 K with the hydrogen pressure of 1.013×105Pa.The ΔG at 77 and 300 K are 37.3 and 301.5 kJ·mol-1,respectively.The positive ΔG indicates that high hydrogen pressure is necessary to make hydrogen hold on B12Sc4.As done by Zhao et al.6,the influence of hydrogen pressure can be estimated with ideal gas model for hydrogen.Our calculation shows that B12Sc4-12H2will release hydrogen when the hydrogen pressure drops back below 129.696×105Pa at 77 K.At 300 K,it needs a drastically high pressure to hold the hydrogen on B12Sc4.It should be noted that the ideal gas model can only give a gross estimation,especially at high pressure.So,the real pressure of hydrogen should be lower than that estimated by ideal gas model.For B12Ti4-8H2,at 300 K it will release hydrogen when the hydrogen pressure drops back below 0.02×105Pa.However,it is difficult to release the last hydrogen molecule.It is also hard to release the hydrogen at 77 K.
4 Conclusions
In the present work,the structures and hydrogen adsorption properties of B12Sc4and B12Ti4clusters were investigated with the first-principles calculations.Both in B12Sc4and B12Ti4,metal atoms prefer binding to B atoms other than clustering together.The B12Sc4can bind up to 12 H2molecules with an ABE/H2of-10.4 kJ·mol-1,while B12Ti4can only host 8 H2molecules at most with an ABE/H2of-50.3 kJ·mol-1.Indeed,the hydrogen adsorption capacities of B12Sc4and B12Ti4clusters we proposed here are not more excellent than a chained B5Ti structure suggested by other stuffs.However,our works would be useful for guiding the design of the hydrogen storage materials based on transition metal-boron clusters.It is very interesting that we find that in B12Sc4,B3rings have s-,p-,and d-like orbitals when interacting with Sc atom.We also find that the Kubas interaction in B12Ti4-nH2complex is much stronger than that in B12Sc4-nH2complex.
Supporting Information Available: The geometries of some calculated B12Sc4and B12Ti4isomers and their relative energies have been included.The detail configures for molecular dynamic(MD)calculation also have been listed.Two pieces of movies demonstrating the MD trajectory of B12Sc4and B12Ti4have been provided.This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Schlapbach,L.;Züttel,A.Nature 2001,414,353.doi:10.1038/ 35104634
(2) Coontz,R.;Hanson,B.Science 2004,305,957.doi:10.1126/ science.305.5686.957
(3) Crabtree,G.W.;Dresselhaus,M.S.;Buchanan,M.V.Phys. Today 2004,57,39.doi:10.1063/1.1878333
(4) Tao,Z.L.;Peng,B.;Liang,J.;Cheng,F.Y.;Chen,J.Meter. China 2009,28,7. [陶占良,彭 博,梁 静,程方益,陈 军.中国材料进展,2009,28,7.]
(5) Xu,W.;Tao,Z.L.;Chen,J.Prog.Chem.2006,18,2.[许 炜,陶占良,陈 军.化学进展,2006,18,2.]
(6) Zhao,X.B.;Xiao,B.;Fletcher,A.J.;Thomas,K.M.J.Phys. Chem.B 2005,109,8880.
(7) Qu,D.Chem.Eur.J.2008,14,1040.doi:10.1002/chem. 200701042
(8) Zaluska,A.;Zaluski,L.;Ström-Olsen,J.O.Appl.Phys.A 2001, 72,157.doi:10.1007/s003390100783
(9) Bogdanovic,B.;Schwickardi,M.J.Alloy.Compd.1997,253,1. doi:10.1016/S0925-8388(96)03049-6
(10) Orimo,S.;Nakamori,Y.;Eliseo,J.R.;Zuttel,A.;Jensen,C.M. Chem.Rev.2007,107,4111.doi:10.1021/cr0501846
(11)Ning,H.;Tao,X.M.;Wang,M.M.;Cai,J.Q.;Tan,M.Q.Acta Phys.-Chim.Sin.2010,26,2267.[宁 华,陶向明,王芒芒,蔡建秋,谭明秋.物理化学学报,2010,26,2267.]doi:10.3866/ PKU.WHXB20100828
(12) Li,G.X.;Chen,X.W.;Bai,J.D.;Lan,Z.Q.;Guo,J.Acta Phys.-Chim.Sin.2010,26,1448.[黎光旭,陈晓伟,白加栋,蓝志强,郭 进.物理化学学报,2010,26,1448.]doi:10.3866/ PKU.WHXB20100540
(13)Wang,H.;Gao,Q.;Hu,J.J.Am.Chem.Soc.2009,131,7016. doi:10.1021/ja8083225
(14) Miao,Y.L.;Sun,H.;Wang,L.;Sun,Y.X.Acta Phys.-Chim. Sin.2012,28,547.[苗延霖,孙 淮,王 琳,孙迎新.物理化学学报,2012,28,547.]doi:10.3866/PKU.WHXB201112301
(15)Yang,Z.;Xia,Y.;Robert,M.J.Am.Chem.Soc.2007,129, 1673.doi:10.1021/ja067149g
(16)Koh,K.;Wong-Foy,A.G.;Matzger,A.J.J.Am.Chem.Soc. 2009,131,4184.doi:10.1021/ja809985t
(17) Zhao,D.;Daren,J.T.;Yuan,D.;Zhou,H.C.Accounts Chem. Res.2011,44,123 and references therein.doi:10.1021/ ar100112y
(18)Zhao,Y.;Kim,Y.H.;Dillon,A.C.;Heben,M.J.;Zhang,S.B. Phys.Rev.Lett.2005,94,155504.doi:10.1103/PhysRevLett. 94.155504
(19) Yildirim,T.;Ciraci,S.Phys.Rev.Lett.2005,94,175501.doi: 10.1103/PhysRevLett.94.175501
(20) Kubas,G.J.J.Organomet.Chem.2001,635,37.doi:10.1016/ S0022-328X(01)01066-X
(21) Sun,Q.;Wang,Q.;Jena,P.;Kawazoe,Y.J.Am.Chem.Soc. 2005,127,14582.doi:10.1021/ja0550125
(22) Sun,Q.;Jena,P.;Wang,Q.;Marquez,M.J.Am.Chem.Soc. 2006,128,9741.doi:10.1021/ja058330c
(23) Wang,Q.;Sun,Q.;Jena,P.;Kawazoe,Y.J.Chem.Theory Comput.2009,5,374.doi:10.1021/ct800373g
(24) Chandrakumar,K.R.S.;Ghosh,S.K.Nano Lett.2008,8,13. doi:10.1021/nl071456i
(25) Liu,W.;Zhao,Y.H.;Li,Y.;Jiang,Q.;Lavernia,E.J.J.Phys. Chem.C 2009,113,2028.doi:10.1021/jp8091418
(26) Rabilloud,F.J.Phys.Chem.A 2010,114,7241.doi:10.1021/ jp103124w
(27) Meng,S.;Kaxiras,E.;Zhang,Z.Nano Lett.2007,7,663.doi: 10.1021/nl062692g
(28) Zhao,Y.F.;Lusk,M.T.;Dillon,A.C.;Heben,M.J.;Zhang,S. B.Nano Lett.2008,8,157.doi:10.1021/nl072321f
(29) Li,Y.C.;Zhou,G.;Li,J.;Gu,B.L.;Duan,W.H.J.Phys.Chem. C 2008,112,19268.doi:10.1021/jp807156g
(30) Wu,G.;Wang,J.L.;Zhang,X.;Zhu,L.J.Phys.Chem.C 2009, 113,7052.doi:10.1021/jp8113732
(31) Li,M.;Li,Y.;Zhou,Z.;Shen,P.;Chen,Z.Nano Lett.2009,9, 1944.doi:10.1021/nl900116q
(32) Zhao,J.;Wang,L.;Li,F.;Chen,Z.J.Phys.Chem.A 2010,114, 9969.doi:10.1021/jp1018873
(33) Li,F.;Zhao,J.;Chen,Z.Nanotechnology 2010,21,134006. doi:10.1088/0957-4484/21/13/134006
(34) Wu,H.S.;Qin,X.F.;Xu,X.H.;Jiao,H.;Schelyer,P.v.R. J.Am.Chem.Soc.2005,127,2334.doi:10.1021/ja046740f
(35) Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/ 1.464913
(36) Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B 1988,37,785.
(37) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision C.01;Gaussian Inc.:Pittsburgh,PA,2004.
(38) Zhao,Y.;Truhlar,D.G.J.Chem.Theory Comput.2005,1,415. doi:10.1021/ct049851d
(39) Mohan,N.;VIjayalakshmi,K.P.;Koga,N.;Suresh,C.H. J.Comput.Chem.2010,31,2874.
(40) Boustani,I.Phys.Rev.B 1997,55,16426.doi:10.1103/ PhysRevB.55.16426
(41) http:/cp2k.berlios.de.
(42) Balasubramanina,K.Chem.Phys.Lett.1987,135,288.doi: 10.1016/0009-2614(87)85158-8
(43) Bauschlicher,C.W.,Jr.;Walch,S.P.J.Chem.Phys.1982,76, 4560.doi:10.1063/1.443532
(44) Thomas,J.R.;Quelch,G.E.;Seidl,E.T.;Schaefer,H.F.,III. J.Chem.Phys.1992,96,6857.doi:10.1063/1.462575
(45) Lukens,W.W.,Jr.;Matsunaga,P.T.;Andersen,R.A. Organometallics 1998,17,5240.doi:10.1021/om980601n
(46) Pattiasina,J.W.;Bolhuis,F.;Teuben,J.H.Angew.Chem.Int. Edit.1987,26,330.doi:10.1002/anie.198703301
January 15,2012;Revised:May 14,2012;Published on Web:May 15,2012.
Hydrogen Storage Properties of B12Sc4and B12Ti4Clusters
MALi-Juan WANG Jian-Feng JIAJian-Feng*WU Hai-Shun
(School of Chemistry and Materials Science,Shanxi Normal University,Linfen 041004,Shanxi Province,P.R.China)
The structures and hydrogen storage properties of two stable B12Sc4and B12Ti4clusters have been investigated using ab initio calculations.No metal atom clustering occurs in the clusters.The B12Sc4hosts 12 H2to achieve 7.25%(mass fraction)hydrogen storage capacity with an average binding energy (ABE)of-10.4 kJ·mol-1per H2,while the B12Ti4can only host 8 H2(4.78%,mass fraction)with a higher ABE (-50.2 kJ·mol-1per H2).High hydrogen pressure is needed for B12Sc4to hold 12 H2,even at 77 K. Electronic structure analysis indicates that the Kubas interaction in the B12Ti4-nH2complex is much stronger than that in the B12Sc4-nH2complex.
Boron cluster;Metal doping;Hydrogen storage;Adsorption;Ab initio calculation
10.3866/PKU.WHXB201205151
∗Corresponding author.Email:jjf_sxtu@yahoo.com.cn;Tel:+86-357-2051375.
The project was supported by the National Basic Research 973 Pre-research Program of China(2010CB635110)and Natural Science Foundation of Shanxi Province,China(2010011012-2).
973计划前期研究专项课题(2010CB635110)与山西省自然科学基金(2010011012-2)资助项目
O641
