脂质沉积性肌病的临床和病理特点
2012-11-20笪宇威贾建平
袁 泉 王 敏 卢 岩 刘 璐 笪宇威 贾建平
脂质沉积性肌病 (lipid storage myopathy,LSM)是由于影响脂质代谢的一些酶或肉毒碱等缺乏,导致异常的脂滴在肌纤维细胞内的聚集,临床以四肢近端为主的肌无力、萎缩为特点。1972 年Engel等报道了首例[1],本病临床表现十分复杂,容易与炎症性肌病、进行性肌营养不良混淆,故现将于我科就诊的5例患者的临床资料综合分析如下。
1 临床资料
1.1 一般资料 2004年1月~2009年12月在本科就诊的LSM 患者5例,男2例,女3例;发病年龄16~46 岁;5 例患者均无相关家族史。5 例LSM 患者的主要临床资料见表1。
典型病例:女,19岁。9岁时出现四肢力弱,行走费力,进行性加重,渐出现持物及行走困难,曾于外院就诊为“肌营养不良”,对症治疗后症状有所改善后出院。4个月前四肢无力加重,1周来渐出现咀嚼无力,吞咽困难,言语欠清及呼吸费力,肌电图检查示肌源性损害,为进一步明确诊断,行肌肉活检。入院查体:神志清楚,言语欠清,颅神经未见异常,双上肢肌力近端Ⅲ级、远端Ⅳ级,双下肢肌力Ⅳ级,四肢肌张力低,腱反射减低,深、浅感觉正常,四肢肌肉有轻度压痛及萎缩。肌电图检查示肌源性损害,血清肌酸磷酸肌酶(CK)9547 IU/L,乳酸脱氢酶(LDH)1304 IU/L。左肱二头肌活检示:HE 染色在萎缩肌纤维和部分正常肌纤维中可见大量脂质沉积,呈筛孔状小空泡,未见变性、坏死纤维,无炎性细胞浸润,肌间质亦无炎性细胞浸润(图1);ORO染色可见空泡呈红染,提示Ⅰ型肌纤维中有大量脂质颗粒沉积空泡(图2),PAS染色未见明显异常,ATP酶染色Ⅰ型和Ⅱ型肌纤维呈棋格样分布,萎缩和含大量脂滴的肌纤维主要是Ⅰ型和部分Ⅱa型肌纤维亦受累。患者入院初诊断为进行性肌营养不良,行肌肉活检后病理诊断为脂质沉积性肌病,采用激素、辅酶Q10、B 族维生素、限制脂肪摄入量治疗后患者症状好转。
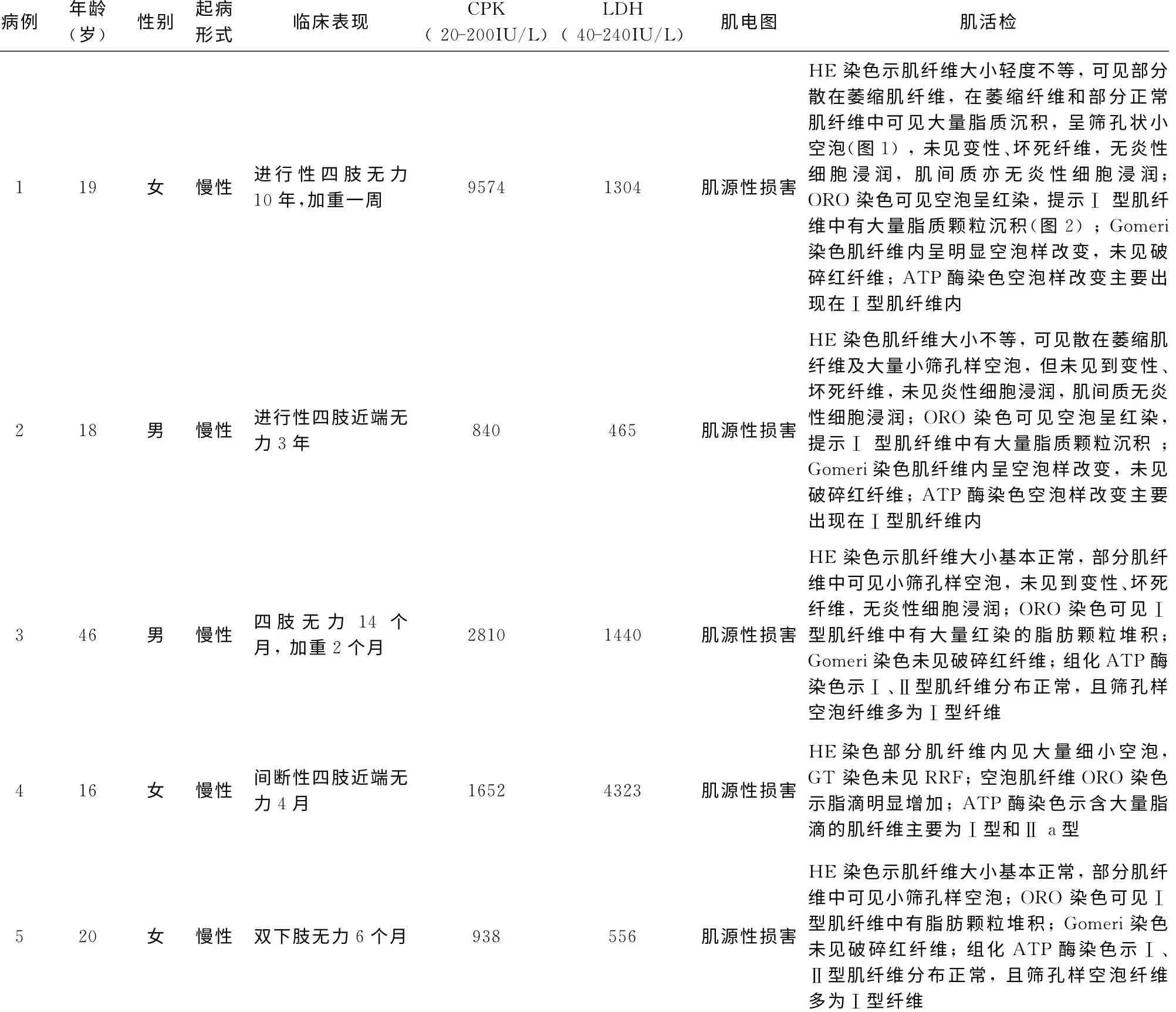
表1 5例LSM 患者临床资料
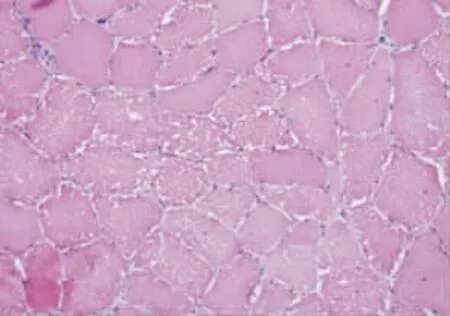
图1 部分肌纤维内细小空泡伴裂隙形成(HE×400)
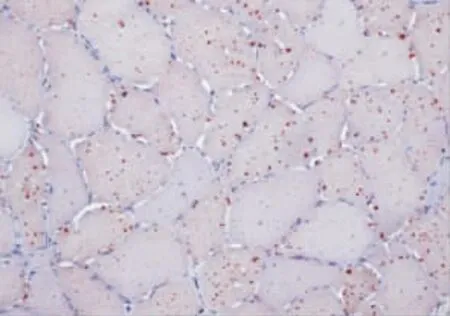
图2 部分肌纤维内脂滴明显增加(ORO×400)
1.2 治疗与转归
患者确诊后给予激素、辅酶Q10、B 族维生素、限制脂肪摄入量,进食高碳水化合物等治疗2-3周,患者肌无力症状渐改善,血清肌酶基本恢复正常;例5同时予以补充肉碱等治疗,6 d后无力症状逐渐好转,血清肌酶各项指标均趋于正常。
2 讨 论
脂质沉积性肌病临床多见于青少年,亦可见于成年人。本病是由于影响脂质代谢的线粒体酶或肉碱缺乏,致使脂肪酸无法进行氧化代谢产生能量,干扰了肌纤维内脂质代谢,导致肌细胞内脂肪堆积而引起的肌病[2],1972年Engel等人首次对脂质沉积性肌病进行了报道。依据脂肪酸代谢通路的各种生化异常,可将脂质沉积性肌病分为肌肉肉碱缺乏症、系统性肉碱缺乏症、肉碱棕榈酰基转移酶缺乏症和线粒体内其他酶缺乏引起的继发性肉碱缺乏,如乙酰辅酶A 脱氢酶缺乏症、肌腺苷脱氨酶、细胞色素C氧化酶缺乏症等[3,4]。
肌肉肉毒碱缺乏症:多见于儿童或青少年,亚急性或慢性起病;患者主要表现为进行性肌无力,以近端肢体为甚,疲劳不耐受,劳累后加重,可伴有肌痛,该病变局限于肌肉,一般不累及肾脏和肝脏。实验室检查可见骨骼肌肉碱水平低,血浆肉碱水平正常,肝脏或心脏的肉碱水平正常,口服肉碱治疗临床症状可中度改善,甚至完全恢复正常[5,6]。本组5例患者符合此型特点,4例青少年起病,1例成年起病,近端肌肉无力为主,运动不耐受,病程有波动性,2例患者入院后应用激素治疗好转,另3例患者未用激素,给予低脂高碳水化合物饮食、维生素、肉碱补充、能量合剂、辅酶Q10治疗取得良好效果。而原发性系统性肉碱缺乏,发病年龄大多为儿童,甚至婴儿,进行性全身无力,常伴有肌肉、肝脏、脑、肾脏等多个脏器的损害,血浆和多种组织中肉碱水平下降,血清肌酶谱升高,肌电图肌源性损害,此型患者口服肉碱治疗效果好[7]。
肉碱棕榈酰基转移酶缺乏症发病年龄多为儿童和青少年,主要表现为发作性肌肉疼痛、无力和发作性肌红蛋白尿,发作时肌酶增高,发作间歇期无任何症状,肌电图正常,肌肉活检也可以正常或有轻度脂肪堆积。给予卡尼汀治疗无效,给予中链脂肪酸、低脂肪高糖饮食、预防感染治疗以及避免重活动和饥饿可减少肌红蛋白尿发作[8,9]。
此外,线粒体内其他酶缺乏,包括乙酰辅酶A脱氢酶缺乏症、肌腺苷脱氨酶、细胞色素C 氧化酶缺乏症等缺乏,可引起继发性肉碱或肉碱棕榈酰基转移酶缺乏和脂肪氧化障碍,导致脂肪沉积,而此种病因导致的脂质沉积性肌病属于线粒体肌病的范畴[7,10]。
综上所述,脂质沉积性肌病的临床表现为肌肉无力萎缩,近端无力明显,可伴有构音不清、咀嚼无力、抬头无力等症状,肌酶指标增高,肌电图可见肌源性损害,肌活检发现肌纤维有空泡样改变,脂肪染色脂滴增多即可确诊。鉴别诊断方面主要与多发性肌炎、肢带型进行性肌营养不良、重症肌无力等鉴别,主要依靠肌肉活检鉴别。该病预后较好,治疗采用低脂饮食,B 族维生素,肉碱补充结合能量支持。对肌肉肉毒碱缺乏患者可用肾上腺皮质激素治疗,通常有效。临床医师需提高对LSM 的认识,熟悉本病的临床特点,以期早期确诊治疗、预防并发症及改善患者生活质量。
1 Engel AG,Seikert RG.Lipid storage myopathy responsive to prednisone.Arch Neruol,1972,27(2):174-181.
2 Victor M ,Ropper AH.Adams and victor’s principles of neurology.7th New York:McGraw-Hill,2001,1512-1528.
3 Vockley J,Whiteman DA.Defects of mitochondrial-oxidation:a growing group of disorders.Neuromuscul Disord,2002,12(3):235-246.
4 Bertorini T,Yeh YY,Trevisan C,et al.Carnitine palmitl transferase deficiency:myoglobinuria and espiratory failure.Neurology,1980,30(3):263-271.
5 笪宇威,贾建平.原发性肉碱缺乏致脂质沉积性肌病的临床与病理特点.临床神经病学杂志,2007,20(3):191-193.
6 陈兴泳,胡 伟,汪银洲等.脂质沉积性肌病临床特征.中国神经精神疾病杂志,2008,34(11):675-676.
7 卫 华,王玉平.脂质沉积性肌病.脑与神经疾病杂志,2005,13(2):141-142.
8 徐德民,武 涛,谢卫龙,等.脂质沉积性肌病临床、神经电生理及肌肉病理研究.中国神经免疫学和神经病学杂志,2009,16(4):259-2 61.
9 丁 玲,毕方方,梁静慧,等.3例脂质沉积性肌病临床与病理分析.卒中与神经疾病,2006,13(2):120-122.
10 Turnbull DM,Shepherd IM,Sherratt HS,et al.Lipid storage myopathy associated with low acyl-CoA dehydrogenase activities.Brain,1988,111(Pt 4):815-828.
