高效液相色谱法测定18F-FDG 注射液中氨基聚醚含量
2012-11-12王晓静叶肇云赵秀岩
赵 岩,王晓静,叶肇云,赵秀岩,姜 华
(原子高科股份有限公司,北京 102413)
氟[18F]脱氧葡萄糖(18F-FDG)注射液是目前应用最广泛的正电子发射断层扫描(PET)显像剂,主要用于恶性肿瘤的诊断和心肌及大脑葡萄糖代谢的测定。氨基聚醚(K2.2.2)是制备18FFDG 的重要试剂,由于具有较强的毒性,其含量是18FDG注射液质量控制中的一项重要指标。国内外有关K2.2.2含量测定方法较多,主要有薄层色谱法[1]、液相色谱法[2]、液质联用色谱法[3-4]、气相色谱法[5]和紫外分光光度法[6],其中,薄层色谱法为半定量法,不能准确测定K2.2.2,液相色谱法、气相色谱法和紫外分光光度法的检测限均大于0.25mg/L,液质联用色谱法灵敏度最高,可以精确检测到20μg/L,但因仪器昂贵不易推广。目前,我国18FDG 注射液的生产中依照的标准为国家药品监督管理局颁布的18F-FDG 注射液试行国标,检测限为K2.2.2低于25mg/L,检测方法是由赵贵植等[6]建立的紫外分光光度法,利用K2.2.2在pH 6.4柠檬酸-氢氧化钠缓冲溶液中与铅(Ⅱ)形成的络合物,在紫外波长253nm 处进行定量测定。该方法中本底溶液对结果有干扰,使测定值比实际值偏高,样品用量较大,需使用0.5mL样品完成一次测定。因此,亟需建立准确度更高和样品用量更少的检测方法。
高效液相色谱法具有灵敏度高、样品用量少,可分离混合物的优点。因此,本工作拟在文献[2]的基础上,采用高效液相色谱法对18FFDG 注射液中K2.2.2含量进行测定,优化实验条件,建立适用于本实验室的高效液相色谱测定方法。
1 主要试剂与仪器
1.1 主要试剂
K2.2.2对照品:纯度>99.0%,ABX 公司;脱氧葡萄糖(2-Deoxy-D-glucose),纯度>99.5%:Sigma公司。甲醇、乙腈:国产色谱纯;磷酸三铵、乙酸铵:国产分析纯
1.2 主要仪器
高效液相色谱(HPLC)仪:XTerraRP18 色谱柱(150mm×3.9mm,5μm),美国Waters公司;UV-2450紫外可见分光光度计:岛津公司;AB135-S电子天平:瑞士Mettler公司。
2 实验方法
2.1 HPLC流动相的确定
分别采用50%乙腈、85%乙腈、V(50 mmol/L磷酸铵(pH9.3)∶V(乙腈)=21∶4、V(50 mmol/L 磷酸铵(pH 8.3))∶V(乙腈)=21∶4和V(50 mmol/L 乙酸铵)∶V(乙腈)=1∶1等5 种不同组分的流动相对100 mg/L K2.2.2和0.5g/L 脱氧葡萄糖进样分析。确定最佳流动相。
2.2 HPLC流速的影响
采用V(50 mmol/L乙酸铵)∶V(乙腈)=1∶1为流动相,流速分别为0.5、1.0mL/min,测定18FDG注射液中K2.2.2的含量,确定最佳流速。
2.3 HPLC测定波长的选择
采用V(50 mmol/L 乙酸铵)∶V(乙腈)=1∶1为流动相,流速0.5 mL/min,向高效液相色谱柱中加入10μL 100mg/L的K2.2.2溶液,分别于210、215、220nm 波长处进行检测,确定最佳检测波长。
2.4 工作曲线
在优化的色谱条件下,分别精密量取10μL,浓度为1、10、20、30、50mg/L 的K2.2.2注入液相色谱仪进行分析。计算色谱峰面积,以峰面积为纵坐标,K2.2.2含量为横坐标,制做工作曲线。
2.5 精密度
将12.5mg/L K2.2.2和0.5g/L脱氧葡萄糖混合,为混合样品1;将25mg/L K2.2.2和0.5g/L脱氧葡萄糖混合,为混合样品2。在优化的色谱条件下,分别将混合样品1 和混合样品2 在HPLC仪上连续进样6次,测量K2.2.2含量。计算精密度。
2.6 回收率
在两个混合样品中分别加入0.495 mg 和0.972mg K2.2.2对照品,用HPLC测定样品中K2.2.2的含量,计算回收率。
2.7 测定方法比较
对4批18F-FDG 注射液分别采用紫外分光光度法和HPLC法测定其K2.2.2含量,比较两种方法的测定结果。
3 结果与讨论
3.1 HPLC流动相的确定
对比5种流动相下色谱柱上脱氧葡萄糖的保留和色谱图上K2.2.2峰,可以看出,以V(50 mmol/L乙酸铵)∶V(乙腈)=1∶1 为流动相时,脱氧葡萄糖在色谱柱上无保留,并可检出K2.2.2色谱峰,其余4种流动相在本实验条件下未检出K2.2.2色谱峰。因此,选择V(50mmol/L乙酸铵)∶V(乙腈)=1∶1为流动相。
3.2 HPLC流速的影响
当流速为1.0、0.5mL/min时,K2.2.2的 保留时间分别为1.66min和3.22min,因此,将流速确定为0.5mL/min。
3.3 HPLC测定波长的选择
在吸收波长210、215、220nm 处,K2.2.2吸收峰面积分别为706 707、401 264、147 987。可以看出,检测波长 为210nm 时,K2.2.2吸收峰 面积较大,此时方法的灵敏度较高。因此,确定检测波长为210nm。
3.4 最佳条件下的色谱分析
选用V(50 mmol/L 乙酸铵)∶V(乙腈)=1∶1为流动相,流速为0.5 mL/min,检测波长为210nm,100 mg/L K2.2.2对照品和18F-FDG注射液的HPLC分离结果分别示于图1和图2。由图1和图2可见,K2.2.2对照品和18F-FDG 注射液中测定的K2.2.2保留时间基本一致,说明检测体系适应性较好。

图1 K2.2.2对照品的HPLC谱图
3.5 工作曲线
在选定的HPLC条件下测定K2.2.2样品,得到的工作曲线示于图3。对图3进行线性拟合,线性回归方程为y=265 939.4x+7 490.7,γ=0.999。表明本分析方法在K2.2.21~50mg/L范围内线性良好。检出限为0.5mg/L。

图2 18F-FDG注射液中K2.2.2的HPLC谱图
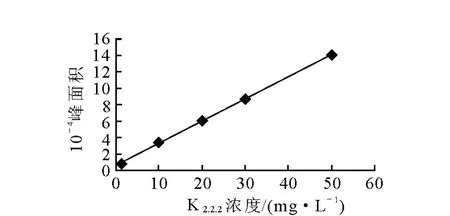
图3 工作曲线
3.6 精密度
精密度结果列于表1。由表1可知,K2.2.2HPLC谱峰面积的精密度小于3.5%。说明该方法精密度良好。

表1 精密度
3.7 回收率
当混合样品中定量加入K2.2.2对照品为0.495、0.972μg时,K2.2.2含量测定值分别为0.475、0.975μg,回收率分别为96.0%、100.3%,表明该方法回收率良好。
3.8 测定方法比较
分别采用紫外分光光度法和HPLC 法对4批18F-FDG 注射液中K2.2.2的含量进行测定,结果列于表2。
紫外分光光度法测定K2.2.2时,由于有干扰物的影响,使测量结果偏高。

表2 两种方法测定结果
4 结论
采用HPLC法可以定量分析18F-FDG注射液中微量的K2.2.2,方法的线性范围为1~50mg/L,γ=0.999,检出限为0.5 mg/L,回收率为96.0%~100.3%。本方法的干扰因素少,与紫外分光光度法相比更准确、灵敏、用样量少,适用 于18F-FDG注射液中微量K2.2.2含 量 的测定。
[1]Chaly T,Dahl JR.Thin layer chromatographic detection of Kryptofix 2.2.2in the routine synthesis of[18F]2-Fluoro-2-deoxy-D-glucose [J].Nucl Med Biol,1989,16:385-387.
[2]Nakao R,Ito T,Yamaguchi M,et al.Simultaneous analysis of FDG,ClDG and Kryptofix 2.2.2in[18F]FDG preparation by high-performance liquid chromatography with UV detection[J].Nucl Med Biol,2008,35:239-244.
[3]Ma Y,Huang BX,Channing MA,et al.Quantification of Kryptofix 2.2.2in 2-[18F]FDG and other radiopharmaceuticals by LC/MS/MS [J].Nucl Med Biol,2002,29:125-129.
[4]张晓军,李云刚,刘健,等.液质联用法测定常用18F药物中Kryptofix 2.2.2的含量[J].同位素,2011,24(3):188-192.
[5]花宁,陈立光.气相色谱法直接测量18F-FDG 中Kryptofix 2.2.2的含量[J].同位素,2007,20(2):105-107.
[6]赵贵植,龙卫忠,李大康,等.分光光度法测定18FFDG 中的K2.2.2[R].北京:原子能出版社,1997.
