慢性铅暴露大鼠海马脑区自噬相关蛋白表达的研究
2012-11-08叶伟峰黄继云廖美华陶蓉蓉张根生楼宜嘉
叶伟峰 ,田 允,黄继云,廖美华,陶蓉蓉,张根生,楼宜嘉,韩 峰
(1.浙江大学医学院附属儿童医院,浙江 杭州 310003;2.浙江大学药理毒理与生化药学研究所,浙江 杭州 310058)
工业及环境来源的铅污染构成对人类长期的危害。铅会通过血脑屏障(blood-brain barrier,BBB)进入中枢神经系统进而产生神经毒性作用。大量研究表明,铅可诱导活性氧的产生,促使细胞和组织产生自由基并导致氧化损伤。众多研究提示,自噬/溶酶体途径(autophagy-lysosome pathway)是真核细胞降解蛋白质及功能失调的细胞器的重要途径,其作为一种防御机制可清除胞质内的细胞器、代谢产物,进行亚细胞水平的重构,在参与维持细胞内环境稳态过程中发挥着重要作用。然而,作为一把双刃剑,同时它也是一种细胞死亡程序诱导细胞主动性死亡,过度的细胞自噬就会引起细胞过量损伤而导致自噬性死亡。有报道认为铅暴露会导致附睾上皮细胞自噬增加[1]。Likholat 等[2]在铅对肺部蛋白水解系统影响的研究中发现,大鼠肺吸入0.01%醋酸铅1 周后组织蛋白酶B(cathepsin B)表达上调,吸入2个月后会大量激活cathepsin B。然而,由于铅的神经毒性机制有许多方面尚不清楚,有关自噬/溶酶体信号在神经毒性方面的研究资料更少,其许多关联性问题有待继续研究。本研究通过对自噬体膜标志性蛋白质的检测,侧重于探索慢性铅暴露条件下海马脑区是否存在着自噬/溶酶体途径的激活。
1 材料与方法
1.1 动物分组与处理 健康成年SD 大鼠,体重雌性220~250 g,雄性250~280 g,清洁级,购自浙江省实验动物中心,动物合格证号为SCXK(浙20080033)。大鼠每天给12 h 的光照,适应性饲养1 周。实验孕鼠随机分成3组,分别为对照组、低剂量铅暴露组(0.5 g/L)和高剂量铅暴露组(2.0 g/L),按组每窝分笼饲养。对照组给予蒸馏水,低剂量组和高剂量组分别给予醋酸铅饮用水,孕鼠自孕16天起自由饮用直至新生仔鼠21天断乳为止[3]。断乳期仔鼠与母鼠分开按组每窝分笼饲养,仔鼠开始饮用与母鼠相同含量的醋酸铅饮用水直至60天成熟期,进行血样与脑组织样取材并测定。各剂量醋酸铅饮用水均为现配现用。
1.2 子鼠血铅浓度与脑组织铅含量测定 将新鲜抗凝血标本与标准品同时用10%硝酸消化后,10 000 r/min 离心5 min,取上清液20μl,加入石墨管,使用石墨炉原子吸收分光光度法上机检测,测定波长为283.3 nm。由标准品作一条铅原子吸光度与浓度的标准曲线,每管标本根据其铅原子的吸光度,在标准曲线上读得相应的血铅浓度。将浓硝酸与高氯酸按体积4∶1配置成混合酸消化液,将1 ml 消化液分别加入各放置新鲜海马脑组织的玻璃试管中,在自控电热消化器上将样本组织加热到120 ℃消化4 h,后升温至260 ℃,消化至完全干燥,冷却后灰分加入去离子水溶解定容成1 ml,摇匀待测。脑组织铅测定使用石墨炉原子吸收分光光度法[4],吸500μl 至上样杯,然后进行上机检测,测定波长为283.3 nm。测定后得到脑组织铅测定值(以μg/dl 表示)换算成脑组织铅含量。
1.3 Western blot 检测海马脑区自噬相关蛋白表达 冻存的海马脑组织参考文献报道方法[5]先加入裂解缓冲液(含有50 mmol/L pH 7.5 Tris-HCl,0.5% Triton X-100,4 mmol/L EGTA,10 mmol/L EDTA,1 mmol/L Na3VO4,30 mmol/L Na2P2O7·10H2O,50 mmol/L NaF,50μg/ml leupeptin,25μg/ml pepstatin A,50μg/ml trypsin inhibitor 和1 mmol/L dithiothreitol)并匀浆,然后4 ℃,15 000×g 离心10 min,取上清待用。用Lowry 试剂盒法对蛋白浓度进行定量。含等量总蛋白的样品采用SDS-PAGE 电泳分离[6]。蛋白上样量为10~20μg/道,SDS-PAGE后使用PVDF 膜,将蛋白在180 mA 电流条件下转膜1 h,膜在用含5% 脱脂奶粉的TBST 封闭液中室温封闭1 h,然后加入各对应的抗体:Beclin 1 抗体(1∶2 000);微管相关蛋白1 轻链3(microtubule-associated protein 1 light chain 3,LC3)抗体(1∶2 000);溶酶体相关膜蛋白2(lysosomal-associated membrane protein 2,LAMP2)抗 体(1∶2 000);β-actin 抗 体(1∶5 000)。4 ℃摇床上孵育过夜,次日用TBST 缓冲液室温在摇床上洗膜3次,每次10 min,后加相应二抗(1∶5 000)摇床上室温孵育膜60 min,然后相同条件下用TBST 缓冲液室温摇动洗膜3次。用ECL 试剂盒使膜上的免疫活性蛋白显色。胶片上蛋白条带经扫描后用Quantity one软件进行光密度分析定量。
1.4 免疫细胞化学法检测cathepsin B 蛋白表达 待检测脑组织样本4%多聚甲醛固定,PBS洗3次,每次5 min;再用2% BSA 封闭1 h,PBS 洗3次,每次5 min;一抗cathepsin B 抗体(monoclonal antibody,1∶200);4 ℃孵育过夜。次日用PBS 洗3次,每次5 min,二抗Alex594标记兔二抗(稀释度1∶200),室温孵育2 h。再用PBS 洗3次,每次10 min。封片后用共聚焦显微镜观察,拍照记录。
1.5 统计学处理 GraphPad Prism 5(GraphPad Software Inc.,San Diego CA)统计分析软件处理所得数据,使用单因素方差分析及Bonferroni 方法作Post-hoc 检验,各计量资料均以平均值±标准差()表示,P<0.05 时表示有统计学意义。
2 结果
2.1 慢性铅暴露后子代大鼠血铅浓度与脑组织铅含量测定 统计分析结果显示,低剂量与高剂量铅暴露组血铅浓度均高于对照组(4.94±1.34)μg/L,分别为(97.83±24.66)μg/L 和(217.72±43.50)μg/L,具有显著性差异(P<0.01,图1A)。与此相一致,海马脑区铅含量测定结果显示,低剂量铅暴露组海马脑铅含量(1.26±0.31)μg/g 与高剂量铅暴露组海马脑铅含量(1.83±0.18)μg/g 明显高于对照组(0.52±0.13)μg/g,差异有显著性(P<0.01,图1B)。

图1 慢性铅暴露后子代大鼠血铅浓度与海马脑组织铅含量的变化Fig.1 Changes of lead content in blood and hippocampus in chronic lead-exposed rats
2.2 慢性铅暴露下子代大鼠海马脑区Beclin 1表达变化 研究结果显示,与对照组相比,子代大鼠海马自噬相关蛋白Beclin 1 表达量在低剂量铅暴露后有明显下调(P<0.05),而高剂量铅暴露组Beclin 1 表达量却显著上调(P<0.05,图2)。
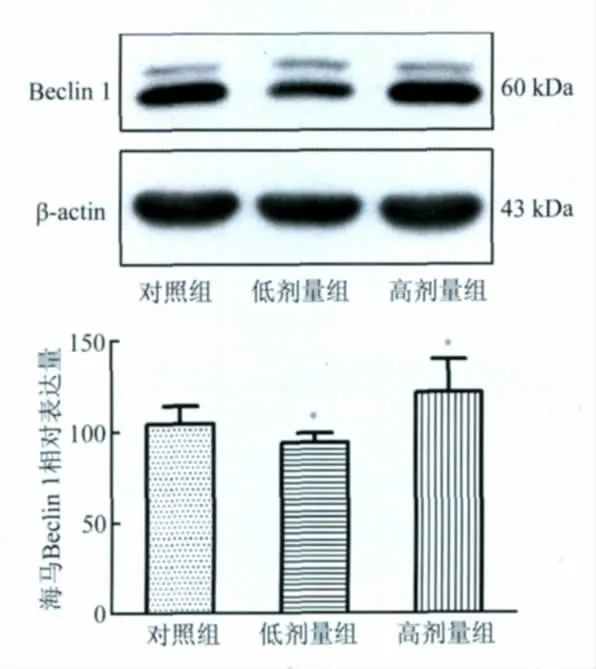
图2 子代大鼠慢性铅暴露后海马Beclin 1 的表达变化Fig.2 Changes of Beclin 1 levels in hippocampus following chronic lead exposure in rats
2.3 慢性铅暴露下子代大鼠海马脑区LC3 表达变化 用Western blot 检测LC3-Ⅱ与LC3-Ⅰ在慢性铅暴露后的表达改变。结果显示,子代大鼠海马LC3-Ⅱ/LC3-Ⅰ比值在低剂量铅暴露后与对照组相比无明显变化,高剂量铅暴露后呈显著上调(P<0.001,图3)。
2.4 慢性铅暴露下子代大鼠海马脑区LAMP2表达变化 Western blot 结果显示,在120 kDa可检测到LAMP2 的条带。子代大鼠在低剂量与高剂量铅暴露后海马自噬相关蛋白LAMP2表达量与对照组相比无明显差异(P >0.05)。

图3 子代大鼠慢性铅暴露后海马LC3-Ⅱ/LC3-Ⅰ的表达变化Fig.3 Changes of LC3 levels in hippocampus following chronic lead exposure in rats
2.5 慢性铅暴露下子代大鼠海马脑区cathepsin B 表达变化 我们采用免疫荧光组织化学技术对cathepsin B 在海马神经元的表达定位进行研究。激光共聚焦显微镜结果显示,慢性铅暴露高剂量组(d、e、f)较正常对照组(a、b、c)的cathepsin B 在海马锥体神经元的阳性表达明显增加,推测慢性铅暴露诱导了cathepsin B 在海马神经元的活性增加(图4)。
3 讨论
铅对儿童脑智力发育及神经行为的神经毒性问题已经受到世界各国学者的高度关注。作为具有典型神经发育毒性的重金属元素,铅对中枢神经系统的损害是不可逆的[7-8]。儿童作为特殊群体,与成人相比铅更易透过血脑屏障。有研究表明,在相同条件下通过血脑屏障的铅含量,儿童是成人的18 倍。铅暴露的年龄越小,对铅神经毒性的易感性越高,血脑屏障阻止铅进入脑内的能力越差[9-10]。此外,据研究显示铅容易透过胎盘屏障,由母体转运胎儿体内,故胎儿的血铅浓度与母体是一致的,铅还可通过乳汁排泄,母体可通过哺乳将铅传递给新生儿。由此可见,母体铅污染对子代产生的影响不容忽视,更需关注的是儿童发育早期铅暴露所产生的智力和神经行为损害在停止铅暴露后可能持续到成年阶段[11-14]。本研究以慢性铅暴露子代大鼠作为研究对象,以自噬/溶酶体信号途径为切入点,系统研究铅暴露对大鼠海马脑区自噬相关蛋白的表达变化,为铅暴露神经毒性分子机制研究提供重要线索。本研究结果提示:慢性铅暴露可诱导大鼠海马脑区发生自噬/溶酶体途径关联信号蛋白表达变化,可能与海马脑区铅含量增加及神经元损伤程度存在内在关联性。

图4 免疫荧光组织化学观察子代大鼠慢性铅暴露后海马锥体神经元cathepsin B的表达变化Fig.4 Changes of cathepsin B immunofluorescence staining in hippocampus following chronic lead exposure in rats
大脑海马区是处理学习、记忆及情感等高级认知功能的重要脑功能区域与信号整合交汇点,可介导神经系统结构与功能的可塑性改变,也是多种外界损伤因素存在时的易损功能单元。本研究选定海马脑区首先检测了不同剂量慢性铅暴露对海马脑区铅含量的影响。现有的研究结果表明,慢性铅暴露可损伤大鼠海马CA1 区和DG 区的长时程增强(long-term potentiation,LTP)和长时程抑制(long-term depression,LTD)的诱导[15-17]。实际上,研究人员应用磁共振脑功能成像技术观察了铅影响儿童脑结构和功能的变化,铅中毒儿童的总智商显著下降,铅严重损伤了儿童脑内海马和前额两个与智力发育相关的重要区域[18]。与慢性铅暴露导致的血铅含量增加相一致,我们的研究发现慢性铅暴露直接导致海马脑铅含量增加,与慢性铅暴露量呈现正相关关系,确认了海马脑区是子代大鼠慢性铅暴露条件下的敏感区域。
自噬在神经元损伤和修复过程发挥着双重效应,本研究采用自噬/溶酶体途径关联蛋白标记物,侧重于了解慢性铅暴露条件下海马脑区是否存在着自噬/溶酶体途径的激活。有研究证实,细胞氧化应激损伤与自噬存在一定的关系,而众多研究发现铅暴露后易导致神经细胞氧化损伤。基于这样的关系,我们通过Western blot 检测慢性铅暴露海马脑区自噬相关蛋白Beclin 1、LC3 与LAMP2 的表达。Beclin 1 是自噬体的标志分子之一,大量研究表明Beclin1 不仅参与自噬体的形成,还可通过调节自噬活性对细胞的发生、发展起着重要作用。首先,本研究发现低剂量铅暴露组导致大脑海马区域Beclin 1 表达降低;相反,高剂量铅暴露组Beclin 1 却表现为表达上调,并没有出现与暴露剂量关联的量效关系的变化。值得关注的是,Beclin l 可能同时参与凋亡和自噬两种程序性细胞死亡过程,有研究表明,凋亡抑制因子bcl-2 和Beclin l 相互作用平衡在两种程序性细胞死亡转换过程中起重要调节作用。Scherz-Shouval等[19]研究认为,活性氧(reactive oxygen species,ROS)是自噬激活的上游信号分子,其可以诱导Beclin 1 的表达增多。Beclin 1 进而通过与磷脂酰肌醇3 磷酸激酶(phosphatidylinositol-3-kinase,PI3K)形成复合物来参与募集胞浆蛋白质用于自噬体膜的形成[20-21],其是自噬体形成的早期过程所必需的,与自噬体形成的启动有关。我们推测低剂量与高剂量铅暴露组导致大脑海马区域Beclin 1 表达的差异,与不同剂量铅暴露导致海马神经元的损伤程度不同,所以导致产生的自噬反应强弱不同有关。
微管相关蛋白1 的轻链3(LC3)是自噬体膜标志性蛋白质。LC3 在体内同时以两种不同分子量的形式存在,其中18 kDa 的LC3 蛋白是微管相关蛋白1 的轻链亚基,称为LC3-Ⅰ;16 kDa 的LC3 蛋白则是自噬体膜的必须组分,称为LC3-Ⅱ,定位于自噬前体和自噬体的内外膜上[22-23]。本研究中,我们检测了慢性铅暴露后LC3-Ⅱ/LC3-Ⅰ的比值变化,结果发现高剂量铅暴露组导致大脑海马区域LC3-Ⅱ/LC3-Ⅰ的比值明显增加,但低剂量铅暴露组未见明显变化。提示慢性高剂量铅暴露导致胞浆可溶型LC3-Ⅰ向胞浆蛋白内膜结合型LC3-Ⅱ的形式转化。因为自噬发生时细胞内LC3-Ⅱ含量会增加,同时LC3-Ⅰ向LC3-Ⅱ形式转化也会明显增多,所以通过监测LC3-Ⅱ/LC3-Ⅰ比值可以了解自噬水平变化,比值升高说明慢性高剂量铅暴露组细胞早期自噬水平增加[24-25]。
自噬过程可将胞质中蛋白质等大分子及生理或病理引起破损的细胞器,在单位膜包裹的囊泡中大量降解,并在溶酶体的参与下进行再循环利用[26]。自噬体可与溶酶体融合形成自噬溶酶体,降解其包裹的内容物,实现细胞自身代谢及一些细胞器的更新。LAMP2 是一种高度糖基化的溶酶体膜内蛋白,可能参与维持溶酶体膜的完整性以及作为一种转运受体协助蛋白进入溶酶体[27],其可以作为溶酶体的标记物[28]。当自噬溶酶体聚集会导致LAMP2 减少并具有相关性,LAMP2 可能对自噬机制有重要作用。但是,我们在慢性铅暴露实验大鼠模型中,未发现慢性铅暴露可诱导LAMP2 的蛋白表达发生变化,这可能与我们所观察的病程(60 d)和脑区相关。Cathepsin B 是溶酶体蛋白酶,属于木瓜蛋白酶家族中的半胱氨酸蛋白水解酶,在正常情况下主要以酶原的形式存在于溶酶体内发挥蛋白水解作用,大约只有10%的酶原可被生理性分泌至胞浆,然后可自身激活或被其它蛋白水解酶水解成活性形式,参与一些生理过程。而当受外界信号刺激的病理状态下,由于细胞损伤导致溶酶体膜通透性增加或者破裂,大量cathepsin B 可渗透到胞浆激活参与降解细胞成分,造成进一步的细胞损害。当自噬体与溶酶体融合过程中,溶酶体内cathepsin B 会释放至胞浆而激活水解,因此cathepsin B 亦可以作为溶酶体的标记物,检测胞浆cathepsin B 的表达可能对辅助判断自噬溶酶体融合有一定的意义[28]。我们发现海马脑区高剂量铅暴露组cathepsin B 在海马锥体神经元的阳性染色明显增加,推测慢性铅暴露诱导了cathepsin B 在海马神经元的活性增加。在后续研究中有必要采用酶联免疫吸附测定LAMP2、cathepsin B 活性或采用透射电镜技术了解慢性铅暴露后海马脑区自噬/溶酶体的变化情况。
鉴于Beclin 1、LC3、LAMP2 及cathepsin B分别处于自噬/溶酶体信号途径的不同环节,Beclin 1、LC3 反映早期自噬水平的变化[29-30],而LAMP2 与cathepsin B 多反映溶酶体激活程度[28,31]。我们的研究表明,慢性铅暴露后在海马脑区自噬不同阶段均出现变化,表明自噬/溶酶体途径的稳态失衡参与了慢性铅神经毒性的整个病理过程。有报道称自噬水平增高可能是一种代偿性的保护效应[32]。就本研究所涉及到的慢性铅暴露模型而言,我们推测大鼠海马脑区自噬水平的增高可能与代偿性效应和神经元损伤两者均相关联。在损伤早期或轻度损伤阶段,自噬水平增高可能是一种代偿性的保护效应,但在自噬超过某一阈值过度激活情况下却会损伤细胞器并促发程序性细胞死亡。我们推测严重铅毒性导致的自噬失代偿可能由以下原因造成:损伤过程中氧化应激造成的自噬失能;各种途径的酶作用底物使得自噬途径超负荷,消耗了必要自噬体;受损的神经元引起的溶酶体内蛋白水解酶活性进行性下降。综上所述,本研究表明了慢性铅暴露模型对神经细胞自噬/溶酶体相关蛋白表达会产生影响,从而可能导致神经细胞自噬水平出现变化,并且不同剂量慢性铅暴露会表现为不同的自噬/溶酶体反应,由此我们推测自噬/溶酶体信号转导途径可能参与了慢性铅暴露致神经毒性分子机制过程。继续深入探讨自噬/溶酶体信号转导途径改变与慢性铅中毒导致的大脑病理改变及神经症状发生机制有重要意义。后续研究进一步围绕慢性铅暴露后不同病理损伤时程的自噬形态学及敏感蛋白标志物的变化,不同病理损伤程度与自噬/溶酶体信号关联性,不同功能脑区的自噬程度等多个层面开展。未来的研究也应该逐步注重自噬有关调控药物联合排铅药物等,用于慢性铅中毒防治的临床开发、应用。
[1]WISZNIEWSKA B,MARCHLEWICZ M,PIASECKA M,et al.Phospholipid content and lamellar structures in the epididymal epithelial cells of rats treated chronically with lead acetate[Pb(Ⅱ)][J].Folia Biol(Krakow),1998,46(3-4):215-224.
[2]LIKHOLAT E A,ANAN'EVA T V,ANTONIUK S V,et al.Proteolytic system in lungs upon inhalation exposure to low doses of lead salts [J].Ukr Biokhim Zh,2000,72(6):84-87.
[3]GILBERT M E,KELLY M E,SAMSAM T E,et al.Chronic developmental lead exposure reduces neurogenesis in adult rat hippocampus but does not impair spatial learning[J].Toxicol Sci,2005,86(2):365-374.
[4]LIM S Y,DOHERTY J D,MCBRIDE K,et al.Lead exposure and (n-3)fatty acid deficiency during rat neonatal development affect subsequent spatial task performance and olfactory discrimination [J].J Nutr,2005,135(5):1019-1026.
[5]HAN F,SHIRASAKI Y,FUKUNAGA K.Microsphere embolism-induced endothelial nitric oxide synthase expression mediates disruption of the blood-brain barrier in rat brain[J].J Neurochem,2006,99(1):97-106.
[6]LAEMMLI U K.Cleavage of structural proteins during the assembly of the head of bacteriophage T4[J].Nature,1970,227(5259):680-685.
[7]LIU J,HAN D,LI Y,et al.Lead affects apoptosis and related gene XIAP and Smac expression in the hippocampus of developing rats [J].Neurochem Res,2010,35(3):473-479.
[8]TANG M,LUO L,ZHU D,et al.Muscarinic cholinergic modulation of synaptic transmission and plasticity in rat hippocampus following chronic lead exposure [J].Naunyn Schmiedebergs Arch Pharmacol,2009,379(1):37-45.
[9]HE Rui-fang,ZHANG Yan,YANG Yan-xu,et al(何瑞芳,张 艳,杨艳旭,等).Changes of lead,zinc,copper,iron and calcium in blood of lead poisoned infantal mice [J].Journal of Applied Clinical Pediatrics(实用儿科临床杂志),2006,21(14):936-937.(in Chinese)
[10]MEYER P A,MCGEEHIN M A,FALK H.A global approach to childhood lead poisoning prevention[J].Int J Hyg Environ Health,2003,206(45):363.
[11]WINNEKE G.Inorganic lead as a developmental neurotoxicant:some basic issues and the Dusseldorf experience [J].Neurotoxicology,1996,17(3-4):565-580.
[12]GILBERT S G,WEISS B.A rationale for lowering the blood lead action level from 10 to 2 microg/dL[J].Neurotoxicology,2006,27(5):693-701.
[13]HUANG Xiao-li,HUANG Ge-ling,CHEN Zhi-hao,et al(黄小丽,黄革玲,陈智浩,等).Detection and analysis of lead concentrations in blood of 3 075 children in Xiamen [J].China Medical Herald(中国医药导报),2010,7(31):131-132.(in Chinese)
[14]CHIODO L M,COVINGTON C,SOKOL R J,et al.Blood lead levels and specific attention effects in young children[J].Neurotoxicol Teratol,2007,29(5):538-546.
[15]RUAN D Y,YAN K F,GE S Y,et al.Effects of chronic lead exposure on short-term and long-term depression in area CA1 of the rat hippocampus in vivo[J].Chemosphere,2000,41(1-2):165-171.
[16]GILBERT M E,LASLEY S M.Developmental lead(Pb)exposure reduces the ability of the NMDA antagonist MK-801 to suppress long-term potentiation (LTP)in the rat dentate gyrus,in vivo[J].Neurotoxicol Teratol,2007,29(3):385-393.
[17]CAO X J,HUANG S H,WANG M,et al.S-adenosyl-L-methionine improves impaired hippocampal long-term potentiation and water maze performance induced by developmental lead exposure in rats [J].Eur J Pharmacol,2008,595(1-3):30-34.
[18]YUAN W,HOLLAND S K,CECIL K M,et al.The impact of early childhood lead exposure on brain organization:a functional magnetic resonance imaging study of language function [J].Pediatrics,2006,118(3):971-977.
[19]SCHERZ-SHOUVAL R,SHVETS E,ELAZAR Z.Oxidation as a post-translational modification that regulates autophagy [J].Autophagy,2007,3(4):371-373.
[20]PATTINGRE S,ESPERT L,BIARDPIECHACZYK M,et al.Regulation of macroautophagy by mTOR and Beclin 1 complexes[J].Biochimie,2008,90(2):313-323.
[21]PATTINGRE S,TASSA A,QU X,et al.Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy[J].Cell,2005,122(6):927-939.
[22]WU J,DANG Y,SU W,et al.Molecular cloning and characterization of rat LC3A and LC3B--two novel markers of autophagosome [J].Biochem Biophys Res Commun,2006,339(1):437-442.
[23]KABEYA Y,MIZUSHIMA N,YAMAMOTO A,et al.LC3,GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation[J].J Cell Sci,2004,117(Pt 13):2805-2812.
[24]SHVETS E,ELAZAR Z.Flow cytometric analysis of autophagy in living mammalian Cells [J].Methods Enzymol,2009,452:131-141.
[25]TANIDA I,UENO T,KOMINAMI E.LC3 conjugation system in mammalian autophagy[J].Int J Biochem Cell Biol,2004,36(12):2503-2518.
[26]KLIONSKY D J,EMR S D.Autophagy as a regulated pathway of cellular degradation [J].Science,2000,290(5497):1717-1721.
[27]NODA T,FUJITA N,YOSHIMORI T.The late stages of autophagy:how does the end begin?[J].Cell Death Differ,2009,16(7):984-990.
[28]WEI J,FUJITA M,NAKAI M,et al.Enhanced lysosomal pathology caused by beta-synuclein mutants linked to dementia with Lewy bodies[J].J Biol Chem,2007,282(39):28904-28914.
[29]RUCK A,ATTONITO J,GARCES K T,et al.The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C.elegans[J].Autophagy,2011,7(4):386-400.
[30]MIZUSHIMA N.Methods for monitoring autophagy[J].Int J Biochem Cell Biol,2004,36(12):2491-2502.
[31]FORTUNATO F,BURGERS H,BERGMANN F,et al.Impaired autolysosome formation correlates with Lamp-2 depletion:role of apoptosis,autophagy,and necrosis in pancreatitis [J].Gastroenterology,2009,137(1):350-360.
[32]CHERRA S J 3RD,DAGDA R K,TANDON A,et al.Mitochondrial autophagy as a compensatory response to PINK1 deficiency [J].Autophagy,2009,5(8):1213-1214.
