TNFα和TNFR1信号通路对小鼠急性肾损伤敏感性影响的研究
2012-10-30黄梁浒张瑞华吴瑾陈建谭建明郑丰
黄梁浒 张瑞华 吴瑾 陈建 谭建明 郑丰
缺血、药物和感染所造成的急性肾损伤严重影响人类健康。虽然急性肾损伤的发病机制很多,但最常见的原因为氧化应激、能量供应减少和炎症等[1-5]。内质网应激是细胞应激的一种重要方式,其发生常因内质网中未折叠蛋白蓄积过多引起[6-7]。目前在哺乳动物已知的3条主要内质网应激通路:(1)胰腺内质网激酶或真核翻译起始因子 2α(pancreatic ER kinase/ eukaryotic translation initiation factor 2α,PERK/eIF2α)[8-9];(2)肌醇所需酶1/X盒结合蛋白 1(inositolrequiring enzyme 1/X-box binding protein 1,IRE1/XBP-1)[10-12];(3)转录激活因子 6(activating transcription factor 6,ATF6) 信 号 通 路[13-15]。eIF2α的激活可减少蛋白质合成,XBP-1可增加细胞内未折叠蛋白的降解,而ATF6能够提高蛋白质折叠。因此,这3条通路均有利于减轻内质网应激,促进细胞恢复正常状态[16-17]。细胞持续处于内质网应激状态可发生炎症、过度氧化应激和细胞死亡[18-21]。因此,慢性内质网应激被认为与动脉粥样硬化、糖尿病以及神经变性等疾病相关[22-24]。研究发现许多参与急性肾损伤的因素例如缺氧、氧自由基增加、糖类和氨基酸供应减少等可促进内质网产生应激反应,因此认为这种应激可能参与急性肾损伤的发病机制[25-28]。衣霉素是一种内质网应激诱导剂,腹腔注射衣霉素后即可导致动物发生急性肾损伤,这与上述假设一致[17-18]。衣霉素是N-乙酰葡糖胺磷酸转移酶的抑制剂,可抑制N-蛋白质糖基化作用的第一步催化反应,衣霉素可诱导未折叠蛋白积聚,造成体内外广泛的内质网应激[16-17]。作为一种促死亡和促炎症因子,肿瘤坏死因子(tumor necrosis factor-alpha, TNFα)可诱导细胞发生内质网应激反应,并主动参与急性肾损伤病理过程[29-32]。本研究拟从TNFα及其受体着手,检测衣霉素诱导下小鼠肾组织内质网应激的状态,并探讨TNFα在肾组织急性内质网应激损伤过程中的作用。
材料与方法
一、材料
C57 小鼠(雌性、3~ 5 月龄)、TNFα、TNFR1、TNFR2基因敲除小鼠和TNFR1R2联合基因敲除小鼠,均购于 Jackson 实验室(Bar Harbor, ME, US)。所有基因敲除小鼠与C57小鼠回交至少6代,小鼠基因分型参照Jackson实验室提供的方法(http://jaxmice.jax.org/protocols),并 以 TNFα、TNFR1、TNFR2基因敲除小鼠和TNFR1R2联合基因敲除小鼠同窝出生的C57小鼠作为对照。小鼠饲养于实验动物中心的无特定病原体环境中,可自由饮食和喝水。所有动物操作程序经过美国西奈山医学院动物管理和使用委员会批准。
衣霉素、顺铂均购自美国Alexis公司,碱性磷酸酶活性检测和血尿素氮检测试剂盒来自美国Teco Diagnostic公司。所有抗体均由以下厂家提供:碱性磷酸酶组化染色用抗体(美国Zymed Laboratories公司)、GRP78抗体和 GRP-94 抗体(美国ABR-Affinity Bioreagents公司)、磷酸化的 eIF2α抗体(美国Stressgen公司)、总 eIF2α抗体(美 国 Bethyl Laboratories公 司)、CHOP 抗 体(美国Alexis Biochemicals公司)、总 RNA 提取试剂盒(美国Promega公司)、定量PCR试剂(美国Roche公司)、兔抗 Annexin Ⅴ流式检测抗体(美国Novus Biologicals公司)、细胞凋亡检测试剂盒(美国Promega公司)。
二、方法
1.小鼠内质网应激的急性肾损伤模型:采用衣霉素作为内质网应激的诱导剂,选择TNFα基因敲除小鼠(n=20)、TNFR1 基因敲除小鼠(n=20)、TNFR2 基 因 敲 除 小 鼠(n=12)以 及TNFR1R2联合基因敲除小鼠(n=12)和 C57小鼠(n=16)腹腔注射(1mg/kg),2~4 d 后处死小鼠,收集尿、血、肾组织和肝组织。另以顺铂(30 mg/kg×3 d)注射 TNFα 基因敲除小鼠和C57小鼠(n=4/组),建立非内质网应激造成的小鼠急性肾损伤模型作为对照。
2.病理学检测和组织免疫化学染色:小鼠麻醉后,用PBS灌注肾组织,4﹪多聚甲醛固定[33]。固定48 h后,PBS清洗组织,用低熔点石蜡包埋,制成4 µm厚的切片,行糖原染色。
由于碱性磷酸酶染色和水通道蛋白2分别是近曲小管和集合管主细胞标志物,对于衣霉素诱导的肾组织,若进行肾近曲小管标志物、集合管标志物和细胞凋亡的联合染色,首先行碱性磷酸酶(红色)染色,然后再行凋亡染色(棕色或深棕);或者先行凋亡染色(棕色或深棕)然后再行水通道蛋白2染色(蓝色)。具体如下:脱腊后,切片置于含碱性磷酸酶底物的反应缓冲液中,室温下孵育30 min,兔抗小鼠水道蛋白2多克隆抗体(1:20 000)孵育切片,然后加入生物素标记的抗兔二抗,加入碱性磷酸酶-抗生物素链菌素复合物,然后滴加碱性磷酸酶底物。凋亡细胞染色首先用蛋白酶 K(20 mg/ml)消化细胞 2.5 min,然后与末端脱氧核苷酸转移酶反应1 h,加入过氧化物酶标记的抗地高辛,经3,3-四盐酸二氨基联苯胺显色,并用苏木素复染细胞核。显微镜下(×400)计数凋亡细胞,每张切片至少计数10个随机视野。
3.RT-PCR和实时定量PCR:采用总RNA试剂盒提取肾皮质总RNA,并按文献报道方法进行反转录[33-34]。采用半定量PCR检测XBP-1 mRNA的表达,退火温度为55 ℃,共进行PCR 35个循环;GAPDH作为内参照,引物参照文献[34]。应用实时定量PCR检测GRP78、GRP94、EDEM1和CHOP mRNA的表达水平,采用β-actin mRNA作为内参照。所有引物序列见表1。

表1 RT-PCR和定量PCR引物
4.免疫印迹:取适量肾皮质,加入含蛋白酶抑制剂、磷酸化酶抑制剂、EDTA的组织蛋白裂解液[33-34]。组织蛋白定量后,按总蛋白量相同或孔制备电泳上样液。电泳后将蛋白转移至PVDF膜上,加入 GRP78抗体(1:1000)、磷酸化的 eIF2α抗体(1:1000)和 CHOP 抗体(1:1000)4℃孵育过夜,加入已结合HRP的羊抗兔或羊抗小鼠抗体(1:5000)室温孵育1h,曝光显影。显影后将抗体洗脱,再次加入 GRP-94 抗体(1:5000)或总 eIF2α抗体(1:5000)孵育,按上述步骤加入二抗、显影。完成所有目的蛋白分子的显影后,洗脱抗体,加入上样对照β-actin的抗体,进行曝光显影。每个蛋白分子至少重复实验2次。
5.近曲小管细胞和远曲小管细胞培养及凋亡检测:小鼠近曲小管细胞系和远曲小管细胞系均来自转基因小鼠,并用猿猴病毒40 T抗原永生化(Nelison博士和Friedman博士惠赠)[35-36],细胞培养于含10﹪胎牛血清的DMEM-LG培养液中。为了检测内质网应激对细胞凋亡的影响,24孔细胞培养板接种1 × 104细胞/孔,梯度增加衣霉素浓度(20~ 200 ng/ml),37℃、5﹪CO2培养 24 h 后,收集贴壁和悬浮细胞进行抗Annexin Ⅴ染色,荧光素标记的二抗孵育,流式细胞仪计数。此外,采用MTT法检测细胞活性。按文献[35]分离TNFR1、TNFR2基因敲除小鼠和C57小鼠的原代近曲小管细胞,接种于 24 孔板(1×104/孔),每孔细胞加入衣霉素处理。24 h后,清洗并收集细胞进行MTT实验。
6.Salubrinal对衣霉素诱导细胞凋亡的影响:为了检测TNFR1基因敲除小鼠近曲小管细胞中eIF2α磷酸化对衣霉素诱导细胞凋亡的影响,在加入衣霉素前2 h用salubrinal(eIF2α磷酸酶抑制剂,20~ 75 mmol/L)对细胞进行预处理[37]。采用免疫印迹检测salubrinal预处理的TNFR1基因敲除小鼠近曲小管细胞中磷酸化eIF2α的表达水平。同时C57和TNFR1基因敲除小鼠近曲小管细胞加入 TNFα(10 ng/ml)进行预处理,免疫印迹检测其对eIF2α 磷酸化的影响。
结 果
一、衣霉素诱导C57小鼠肾组织发生急性内质网应激反应,并造成近曲小管损伤
C57小鼠经腹腔注射衣霉素24 h后,与对照组相比,葡萄糖调节蛋白78(GRP78,内质网应激反应标志物)的mRNA和蛋白表达水平分别升高至5.5倍和3.5倍,同时磷酸化eIF2α蛋白水平升高至 4.1倍,,活化的 XBP1(XBP1-s)mRNA水平增高至 4.7 倍。衣霉素注射 48~ 72 h 后,可见肾小管出现病理性改变,以肾小管细胞空泡形成、染色质凝聚、固缩及碎裂等典型改变为主。同时小鼠肾组织经TUNEL染色后发现,与对照相比,衣霉素诱导后肾皮质区域出现大量细胞凋亡。
由于病理学改变大多位于近曲小管,本研究选取碱性磷酸酶(近曲小管标记物之一)进行免疫组化染色,结果显示:衣霉素作用后的小鼠肾组织,碱性磷酸酶阳性的近曲小管数量明显减少。双重染色结果显示:凋亡细胞定位于碱性磷酸酶阳性的近曲小管区域,而水通道蛋白2阳性的集合管区域凋亡细胞几乎不可见。此外应用 50 ng/ml和 200 ng/ml的衣霉素培养 48 h 后,结果显示为:近曲小管细胞的死亡率分别为31﹪和100﹪,而远曲小管的分别为1﹪和28﹪。
二、衣霉素诱导TNFα基因敲除小鼠产生更严重的急性内质网应激反应和急性肾损伤,但能被腹腔注射TNFα逆转
资料表明,TNFα 密切参与急性肾组织损伤[30-31],因此本研究利用TNFα基因敲除小鼠和C57小鼠,观察内质网应激反应和急性肾损伤。结果显示,与C57小鼠相比,衣霉素注射后的TNFα基因敲除小鼠表现出广泛、严重的内质网应激相关性肾组织损伤,肾组织HE染色后显微镜下可见TNFα基因敲除小鼠肾皮质和皮髓质交界处,超过80﹪的肾近曲小管出现病理改变,而C57小鼠则不到20﹪,且主要位于皮髓质交界处。
与C57小鼠相比,衣霉素诱导后TNFα基因敲除小鼠肾组织碱性磷酸酶阳性的小管数量显著减少,且碱性磷酸酶染色形态不规则。此外,衣霉素注射的TNFα基因敲除小鼠肾组织中凋亡细胞显著增多。双重染色证实凋亡细胞主要集中在C57小鼠和TNFα基因敲除小鼠肾组织的小管细胞,集合管细胞中未见,TNFα基因敲除小鼠肾组织的凋亡细胞数量为C57小鼠的7.5倍。
由于内质网应激通路的失调可导致肝脂肪变性, 采用油红O染色检测衣霉素注射后的TNFα基因敲除小鼠和C57小鼠肾组织脂质沉积状况,结果显示,C57小鼠肾组织染色基本为阴性,而衣霉素注射后的TNFα基因敲除小鼠肾组织,包括肾小球和肾小管均出现广泛着色。而以往报道认为,TNFα缺失是一种肾损伤保护机制,这与本研究实验结果相反[5,30]。
注射衣霉素 24 h 前,用 TNFα(200 ng/只)腹腔注射 TNFα 基因敲除小鼠(n= 8),可显著降低上述出现的严重小管损伤。病理染色显示:衣霉素注射后的TNFα基因敲除小鼠小管损伤区域达80﹪,而预先用TNFα处理后的小鼠小管损伤面积可降低至10﹪。碱性磷酸酶免疫组织化学染色证实提前注射TNFα的小管细胞保护较好。TUNEL染色也显示,衣霉素诱导的TNFα基因敲除小鼠肾组织凋亡广泛,但在提前输注TNFα后,凋亡细胞明显减少。因此,TNFα显著减轻衣霉素诱导的TNFα基因敲除小鼠急性肾损伤。
三、TNFR1基因敲除小鼠出现更严重的急性内质网应激损伤,但TNFR2基因敲除小鼠未出现
由于TNFα的作用由TNFR1和TNFR2所介导,因此本研究探讨了衣霉素诱导后TNFR1、TNFR2基因敲除小鼠和TNFR1R2联合基因敲除小鼠的改变。结果显示:衣霉素注射后对照、TNFR1、TNFR2、TNFR1R2基因敲除小鼠的血尿素氮(BUN)分别为 21.5 ± 1.8、39.8 ± 2.5、18.6 ± 1.7、23.1 ± 3.8。衣霉素诱导的 TNFR1 基因敲除小鼠和TNFR1R2联合基因敲除小鼠肾组织中碱性磷酸酶阳性的小管数量显著减少。与C57小鼠相比,TNFR2基因敲除小鼠的小管损伤轻微,而TNFR1基因敲除小鼠 和TNFR1R2联合基因敲除小鼠在皮质、皮髓质交界处均出现大于80﹪严重小管损伤,并与凋亡细胞增多显著相关。油红O染色证实衣霉素注射的TNFR1基因敲除小鼠和联合TNFR1R2基因敲除小鼠肾组织出现脂质积聚,但在C57和TNFR2基因敲除小鼠未出现类似反应。
四、TNFα和TNFR1基因敲除小鼠肾组织内质网应激失调
由于未折叠蛋白的调节异常可加重肝、肾内质网应激造成的急性损伤[15,17]。本研究探讨是否TNFα、TNFR1基因敲除小鼠和TNFR1R2联合基因敲除小鼠发生的严重肾组织损伤是由内质网应激的异常调节引起。衣霉素注射48 h后,肾组织GRP78 mRNA表达水平显著增加,并且C57和三系基因敲除小鼠无差异。然而,相对衣霉素注射的C57或TNFR2基因敲除小鼠,GRP94 mRNA水平在衣霉素注射后的TNFα、TNFR1基因敲除小鼠和TNFR1R2双基因敲除小鼠增加更多(表2)。衣霉素诱导肾组织活化的XBP-1出现5倍增加,表明已经激活了XBP-1通路;但C57、TNFα-和TNF受体缺陷的小鼠XBP-1-s表达水平相似。另外,EDEM是一个参与未折叠蛋白降解的基因,且为XBP-1-s的作用靶点之一,在注射衣霉素后其mRNA表达水平出现与C57及所有基因缺陷小鼠等幅度的上调。这些数据提示,XBP-1信号通路可能未参与衣霉素诱导的C57和TNFα、TNFR1基因敲除小鼠以及TNFR1R2双基因敲除小鼠不同程度的肾组织损伤。与C57和TNFR2基因敲除小鼠相比,C/EBP同源蛋白(CHOP)mRNA 表达水平在 TNFα、TNFR1基因敲除小鼠和TNFR1R2双基因敲除小鼠出现更高的表达上调。在衣霉素注射24 h前输注TNFα可阻止TNFα基因敲除小鼠CHOP mRNA水平增加。
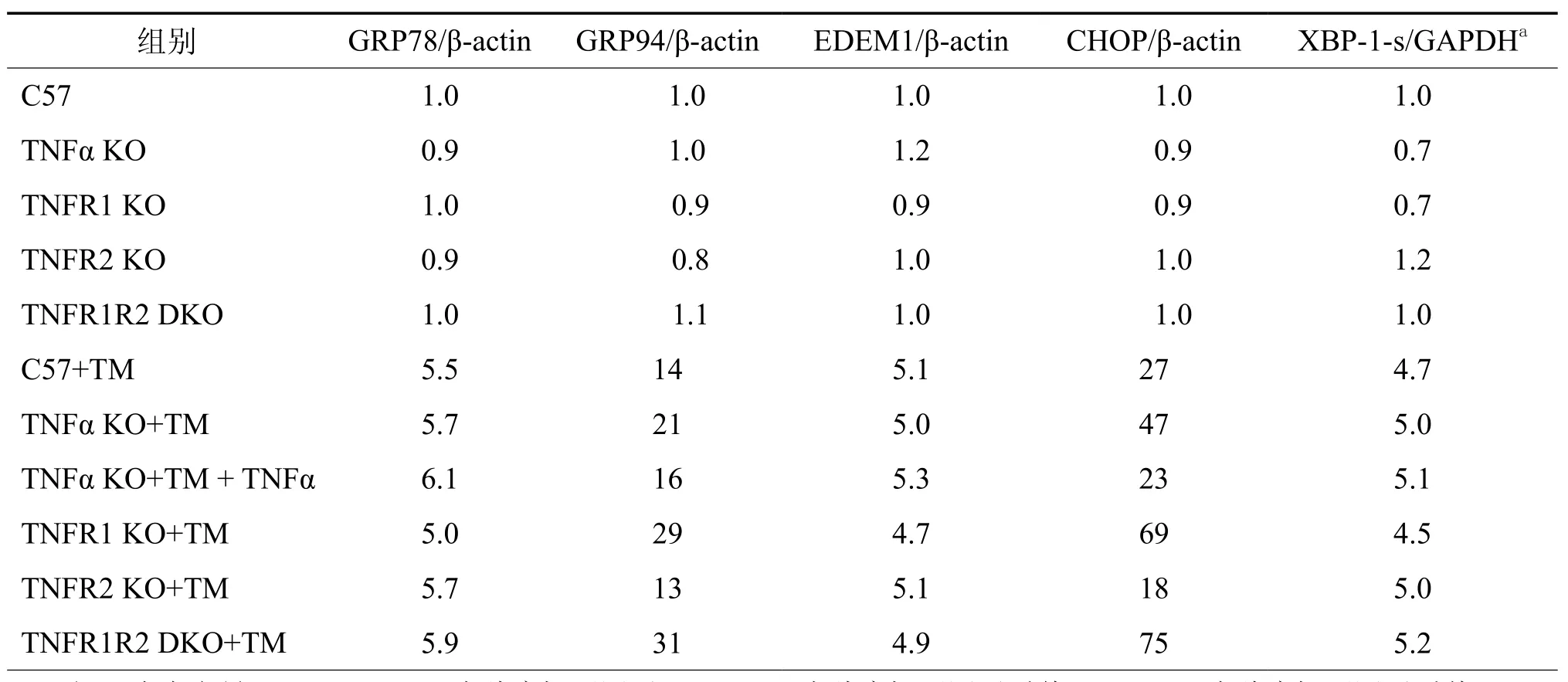
表2 半定量RT-PCR和定量PCR检测内质网相关基因与参照基因的比值
衣霉素注射的所有小鼠均呈现相同幅度的 GRP78 蛋白水平升高。GRP94蛋白水平在衣霉素注射的C57、TNFα基因敲除小鼠、TNFR1基因敲除小鼠和TNFR1R2双基因敲除小鼠的肾组织中表达升高,而TNFR2基因敲除小鼠或提前输注TNFα的TNFα基因敲除小鼠未出现类似反应。衣霉素注射的所有小鼠均出现CHOP蛋白水平增高,虽然TNFR2基因敲除小鼠和提前输注TNFα-的TNFα基因敲除小鼠中衣霉素诱导的肾组织损伤轻微,但在所有基因敲除小鼠CHOP的表达均明显增加,并且不受TNFα的影响。
C57小鼠在衣霉素注射48 h后磷酸化eIF2α表达水平升高,而在衣霉素注射的TNFα和TNFR1基因敲除小鼠的肾组织中未有升高,在衣霉素注射的TNFR1R2双基因敲除小鼠表达最低。TNFα预先输注的TNFα基因敲除小鼠中,在衣霉素作用下大大提高肾组织的eIF2α磷酸化水平。此外,TNFα增加体外近曲小管细胞的eIF2α磷酸化水平。衣霉素对TNFR2基因敲除小鼠肾组织eIF2α磷酸化水平未产生影响。
五、TNFR1基因敲除小鼠近曲小管细胞的eIF2α磷酸化水平降低可导致细胞凋亡增多
从TNFR1、TNFR2基因敲除小鼠和C57小鼠分离出近曲小管细胞,加入衣霉素后,TNFR1基因敲除小鼠的近曲小管细胞死亡较多。为了检测TNFR1基因敲除小鼠细胞死亡是否与内质网应激失调相关,TNFR1、TNFR2基因敲除小鼠和C57小鼠来源的小管细胞加入衣霉素(2 µg/ml)孵育 12 h。结果显示,衣霉素作用下的TNFR1、TNFR2基因敲除小鼠和C57小鼠细胞的 GRP78、XBP-1-s和 EDEM mRNA 的表达水平升高。然而,磷酸化的eIF2α表达水平仅在衣霉素作用下的C57和TNFR2基因敲除小鼠中升高,而未在TNFR1基因敲除小鼠中升高。salubrinal是一种磷酸化eIF2α磷酸酶的化学抑制剂,salubrinal可促进细胞中磷酸化eIF2α表达水平升高[37]。salubrinal(50 mmol/L)可增加 TNFR1基因敲除小鼠和C57小鼠近曲小管细胞的磷酸化eIF2α表达水平。并且salubrinal显著降低衣霉素诱导的TNFR1基因敲除小鼠近曲小管细胞死亡。
讨 论
本研究发现TNFα基因敲除小鼠比C57小鼠更容易引起严重的内质网应激损伤,且这种损伤与广泛的中性脂质蓄积相关。事实上,在衣霉素注射24 h前输注TNFα可完全逆转TNFα基因敲除小鼠肾组织的内质网应激损伤,这说明TNFα可能在保护肾组织免受急性内质网应激方面有直接作用。TNFα 的作用由2个受体介导(TNFR1和TNFR2),TNFR1基因敲除小鼠发生肾衰竭,引起广泛的急性肾损害,包括广泛的凋亡和脂质蓄积;而TNFR2基因敲除小鼠给予衣霉素处理后基本正常;这些结果说明:TNFα对抗内质网应激诱导的小管细胞损伤作用是由TNFR1介导的。
内质网应激可表现为多种形式的急性肾损伤[32,38]。内质网应激通路的失调可导致严重的内质网应激损伤[15,17-18]。例如,XBP-1片段缺失或PERK持续激活可造成内质网应激诱导的细胞凋亡[39-40]。相反,IRE1信号增强可促使细胞抵抗内质网应激诱导的死亡[40]。此外,小鼠缺失ATF6a或PERK可导致内质网应激损伤[9,15]。因此,探讨内质网应激通路的失调是否是TNFα、TNFR1基因敲除小鼠和TNFR1R2双基因敲除小鼠出现内质网应激损伤的原因。在C57小鼠和TNFα、TNFR1、TNFR2基因敲除小鼠中,衣霉素诱导肾损伤时,GRP78 mRNA和蛋白表达水平是相似的;XBP-1-s和EDEM的表达也没有差异。然而,磷酸化eIF2α表达水平却不同,衣霉素诱导下TNFα、TNFR1基因敲除小鼠和TNFR1R2双基因敲除小鼠肾组织中磷酸化eIF2α表达水平下降。如果在衣霉素注射前24 h输注TNFα,则可使TNFα基因敲除小鼠肾组织的eIF2α表达水平再次升高。另外,TNFα可在体外增加近曲小管细胞eIF2α磷酸化的表达水平,这一点也支持TNFα可直接调节eIF2α通路的推测。TNFα可能并不是直接激活PERK而导致eIF2α磷酸化,它可能作用于其他eIF2α激酶,包括双链RNA依赖的蛋白激酶[41]。由于磷酸化eIF2α负调节eIF2B,因此,eIF2的活性降低,蛋白翻译的起始停滞。此外,磷酸化的eIF2α协调多种应激反应通路,并激活抗氧化系统[42]。如果采用基因手段增加eIF2α磷酸化,可增加细胞对抗氧化应激和内质网应激的能力[37,42]。实验结果显示:在衣霉素诱导内质网应激损伤的TNFR1基因敲除小鼠肾组织中,磷酸化的eIF2α表达水平没有明显增加。TNFR1基因敲除小鼠来源的近曲小管细胞的体外实验也证实这一点,表明TNFR1缺失时eIF2α磷酸化对衣霉素呈现无反应性。另一方面,C57或TNFR2基因敲除小鼠的近曲小管细胞反应正常。TNFR1基因敲除小鼠近曲小管细胞的eIF2α磷酸化水平对衣霉素诱导无反应与内质网应激诱导的细胞死亡增加相关。Salubrinal是eIF2α磷酸化酶抑制剂,可增加多种类型细胞的磷酸化eIF2α表达,对抗内质网应激诱导的细胞死亡[37]。Salubrinal同时可增加TNFR1基因敲除小鼠近曲小管细胞中磷酸化eIF2α表达水平,显著降低衣霉素诱导的细胞死亡。结合体内研究,eIF2α通路的失调可加重TNFα和TNFR1基因敲除小鼠急性内质网应激损伤。
TNFα可通过激活NF-κB通路从而保护肾组织免受急性内质网应激诱导的损伤。虽然在诱导内质网应激后,没有发现TNFα或相应受体在肾组织表达的变化,但在内质网应激状态下这个通路可能依然是激活的,因为体内普遍存在循环的TNFα,并且TNFα信号在肾组织内有所增加。内质网应激状态下某些细胞的NF-κB被激活,并诱导TNFα表达[43]。此外,内质网应激可在体外激活TNFR1以及它下游的TNFR2[44–46],虽然这可能具有细胞类型的特异性,但这些结果说明TNFα/TNFR1通路主动参与内质网应激的调节。
已知TNFα可增加顺铂的肾毒性[30],并且多种机制参与顺铂毒性,包括DNA损伤、p21蛋白改变、过多的活性氧分子(ROS)、内质网应激以及炎症[5,30,47]。因此,推测顺铂动物模型中,TNFα的表达水平通常显著升高,它促死亡和炎症的作用将覆盖其对抗内质网应激的保护作用。虽然TNFα可能在缺血再灌注模型中是有害因素,但目前在TNFα及受体缺陷小鼠模型中尚无研究。下一步研究将塑造不同的内质网应激动物模型,以便更好理解内质网应激在急性肾损伤中的作用。
1 Perianayagam MC, Liangos O, Kolyada AY, et al. NADPH oxidase p22 phox and catalase gene variants are associated with biomarkers of oxidative stress and adverse outcomes in acute renal failure[J]. J Am Soc Nephrol, 2007, 18(1):255-263.
2 Brooks C, Wei Q, Cho SG, et al. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models[J]. J Clin Invest, 2009, 119(5):1275-1285.
3 Devarajan P. Update on mechanisms of ischemic acute kidney injury[J]. J Am Soc Nephrol, 2006,17(6):1503-1520.
4 Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease?[J]. Kidney Int, 2004, 66(2):480-485.
5 Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity[J]. J Clin Invest, 2002, 110(6):835-842.
6 Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response[J]. Nat Rev Mol Cell Biol, 2007, 8(7):519-529.
7 Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response[J]. Semin Cell Dev Biol, 2007, 18(6):716-731.
8 Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase[J]. Nature, 1999, 397(6716):271-274.
9 Scheuner D, Song B, McEwen E, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis[J]. Mol Cell, 2001, 7(6):1165-1176.
10 Aragon T, van Anken E, Pincus D, et al. Messenger RNA targeting to endoplasmic reticulum stress signalling sites[J]. Nature, 2009, 457(7230):736-740.
11 Korennykh AV, Egea PF, Korostelev AA, et al. The unfolded protein response signals through high-order assembly of Ire1[J]. Nature, 2009, 457(7230):687-693.
12 Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response[J]. Mol Cell Biol, 2003, 23(21): 7448-7459.
13 Okada T, Yoshida H, Akazawa R, et al. Distinct roles of activating transcription factor 6 (ATF6) and doublestranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response[J]. Biochem J, 2002, 366(Pt 2):585-594.
14 Yoshida H, Matsui T, Hosokawa N, et al. A time-dependent phase shift in the mammalian unfolded protein response[J]. Dev Cell, 2003, 4(2):265-271.
15 Wu J, Rutkowski DT, Dubois M, et al. ATF6-alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress[J]. Dev Cell, 2007, 13(3):351-364.
16 Rutkowski DT, Arnold SM, Miller CN, et al. Adaptation to ER stress is mediated by differential stabilities of prosurvival and pro-apoptotic mRNAs and proteins[J]. PLoS Biol, 2006, 4(11):e374.
17 Rutkowski DT, Wu J, Back SH, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators[J]. Dev Cell, 2008, 15(6):829-840.
18 Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum[J]. Genes Dev, 2004, 18(24): 3066-3077.
19 Malhotra JD, Miao H, Zhang K, et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion[J]. Proc Natl Acad Sci USA, 2008, 105(47):18525-18530.
20 Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease[J]. Cell, 2008, 134(5):743-756.
21 Zhang K, Shen X, Wu J, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response[J]. Cell, 2006, 124(3):587-599.
22 Thorp E, Li G, Seimon TA, et al. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP[J]. Cell Metab, 2009, 9(5): 474-481.
23 Silva RM, Ries V, Oo TF, et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism[J]. J Neurochem, 2005, 95(4):974-986.
24 Song B, Scheuner D, Ron D, et al. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes[J]. J Clin Invest, 2008, 118(10):3378-3389.
25 Sawada N, Yao J, Hiramatsu N, et al. Involvement of hypoxia-triggered endoplasmic reticulum stress in outlet obstruction-induced apoptosis in the urinary bladder[J]. Lab Invest, 2008, 88(5):553-563.
26 Chang SC, Wooden SK, Nakaki T, et al. Rat gene encoding the 78-kDa glucose-regulated protein GRP78: its regulatory sequences and the effect of protein glycosylation on its expression[J]. Proc Natl Acad Sci USA, 1987, 84(3):680-684.
27 Yokouchi M, Hiramatsu N, Hayakawa K, et al. Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response[J]. J Biol Chem, 2008, 283(7):4252-4260.
28 Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities[J]. Nat Rev Drug Discov, 2008, 7(12):1013-1030.
29 Xue X, Piao JH, Nakajima A, et al. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha[J]. J Biol Chem, 2005, 280(40):33917- 33925.
30 Ramesh G, Reeves WB. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure[J]. Am J Physiol Renal Physiol, 2003, 285(4):F610-F618.
31 Meldrum KK, Meldrum DR, Meng X, et al. TNF-alphadependent bilateral renal injury is induced by unilateral renal ischemia-reperfusion[J]. Am J Physiol Heart Circ Physiol, 2002, 282(2):H540-H546.
32 Peyrou M, Hanna PE, Cribb AE. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys[J]. Toxicol Sci, 2007, 99(1):346-353.
33 Zheng F, Plati AR, Potier M, et al. Resistance to glomerulosclerosis in B6 mice disappears after menopause[J]. Am J Pathol, 2003, 162(4):1339-1348.
34 Wu J, Mei C, Vlassara H, et al. Oxidative stress-induced JNK activation contributes to proinflammatory phenotype of aging diabetic mesangial cells[J]. Am J Physiol Renal Physiol, 2009, 297(6):F1622-F1631.
35 Gesek FA, Friedman PA. Calcitonin stimulates calcium transport in distal convoluted tubule cells[J]. Am J Physiol, 1993, 264(4 Pt 2):F744-F751.
36 Wolf G, Neilson EG. Angiotensin II induces cellular hypertrophy in cultured murine proximal tubular cells[J]. Am J Physiol, 1990, 259(5 Pt 2):F768-F777.
37 Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress[J]. Science, 2005, 307(5711):935-939.
38 Pallet N, Bouvier N, Bendjallabah A, et al. Cyclosporineinduced endoplasmic reticulum stress triggers tubular phenotypic changes and death[J]. Am J Transplant, 2008, 8(11):2283- 2296.
39 Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response[J]. Science, 2007, 318(5852):944-949.
40 Lin JH, Li H, Zhang Y, et al. Divergent effects of PERK and IRE1 signaling on cell viability[J]. PLoS One, 2009, 4(1):e4170.
41 Takada Y, Ichikawa H, Pataer A, et al. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation[J]. Oncogene, 2007, 26(8):1201-1212.
42 Lu PD, Jousse C, Marciniak SJ, et al. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2[J]. EMBO J, 2004, 23(1):169-179.
43 Hu P, Han Z, Couvillon AD, et al. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappa B activation and downregulation of TRAF2 expression[J]. Mol Cell Biol, 2006, 26(8):3071-3084.
44 Yang Q, Kim YS, Lin Y, et al. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK[J]. EMBO Rep, 2006, 7(6):622- 627.
45 Mauro C, Crescenzi E, De Mattia R, et al. Central role of the scaffold protein tumor necrosis factor receptorassociated factor 2 in regulating endoplasmic reticulum stress-induced apoptosis[J]. J Biol Chem, 2006, 281(5):2631-2638.
46 Yoneda T, Imaizumi K, Oono K, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress[J]. J Biol Chem, 2001, 276(17):13935-13940.
47 Megyesi J, Safirstein RL, Price PM. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure[J]. J Clin Invest, 1998, 101(4):777- 782.
