烫伤油质量标准研究
2012-10-25王淑华林永强徐丽华
王淑华,林永强,徐丽华
(1.山东省生物药物研究院,山东济南250101;2.山东省食品药品检验所,山东济南250101)
烫伤油是根据中医临床验方研制,由马尾连、紫草、黄芩、地榆、大黄和冰片(合成龙脑)6味中药组成的复方制剂,具有清热解毒,凉血祛腐止痛的功效。1970年批准生产,后陆续收载于《山东省药品标准》1986年版[1]和《中华人民共和国卫生部药品标准—中药成方制剂(第二册)》,标准仅有制法、性状和异物检查项[2]。可控项目较少,为确保制剂质量,对质量标准进行了系统全面的研究,现报道如下。
1 仪器与试药
1.1 仪器 Agilent 6890气相色谱仪;Sartorius CP225D电子天平。
1.2 试药 对照品及对照药材均由中国药品生物制品检定所提供,批号分别为:紫草:110736-200525;黄芩素:111595-200905;大黄素甲醚:110758-200611;大黄酚:110796-200716;冰片:110743-200504;水杨酸甲酯:707-200107。样品:3家生产企业的10批烫伤油批号见表2。氢氧化钠、盐酸、三氯甲烷和乙酸乙酯等均为分析纯。
2 薄层色谱鉴别
本品辅料为麻油,按传统方法用甲醇、乙醇、三氯甲烷等溶剂提取时,均会溶解大量的麻油,很难去除,影响了薄层效果。由于紫草的主要成分为萘醌类化合物,黄芩所含的主要成分黄芩苷、黄芩素和汉黄芩素等均为黄酮类化合物,大黄所含的大黄素、大黄素甲醚和大黄酚等均为蒽醌类化合物,这三类化合物均有一共同的特点:含多个酚羟基,具一定的酸性。因此可用酸碱处理的方法同时从烫伤油中提取紫草、黄芩和大黄三种药材的多酚羟基成分,再用三氯甲烷提取酸性水溶液,即可得到供试品溶液。
2.1 紫草的薄层色谱鉴别 取本品20 g,用2%氢氧化钠溶液20 mL振摇提取,提取液用稀盐酸调节pH值至酸性,用三氯甲烷振摇提取2次,每次20 mL,合并三氯甲烷液,蒸干,残渣加乙醇1 mL使溶解,作为供试品溶液。取紫草对照药材0.5 g,用麻油13 g浸泡24 h后炸至枯黄,滤过,同供试品溶液的制备方法制成对照药材溶液。同时取处方中除去紫草的其他药味,按处方比例制成阴性对照样品,同法制成阴性对照溶液。照薄层色谱法(《中国药典》2010年版一部附录Ⅵ B)试验,吸取供试品溶液、对照药材溶液各10 μL,分别点于同一硅胶G薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。氨蒸气中熏至斑点显色清晰,置日光下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性对照无干扰,见图1。
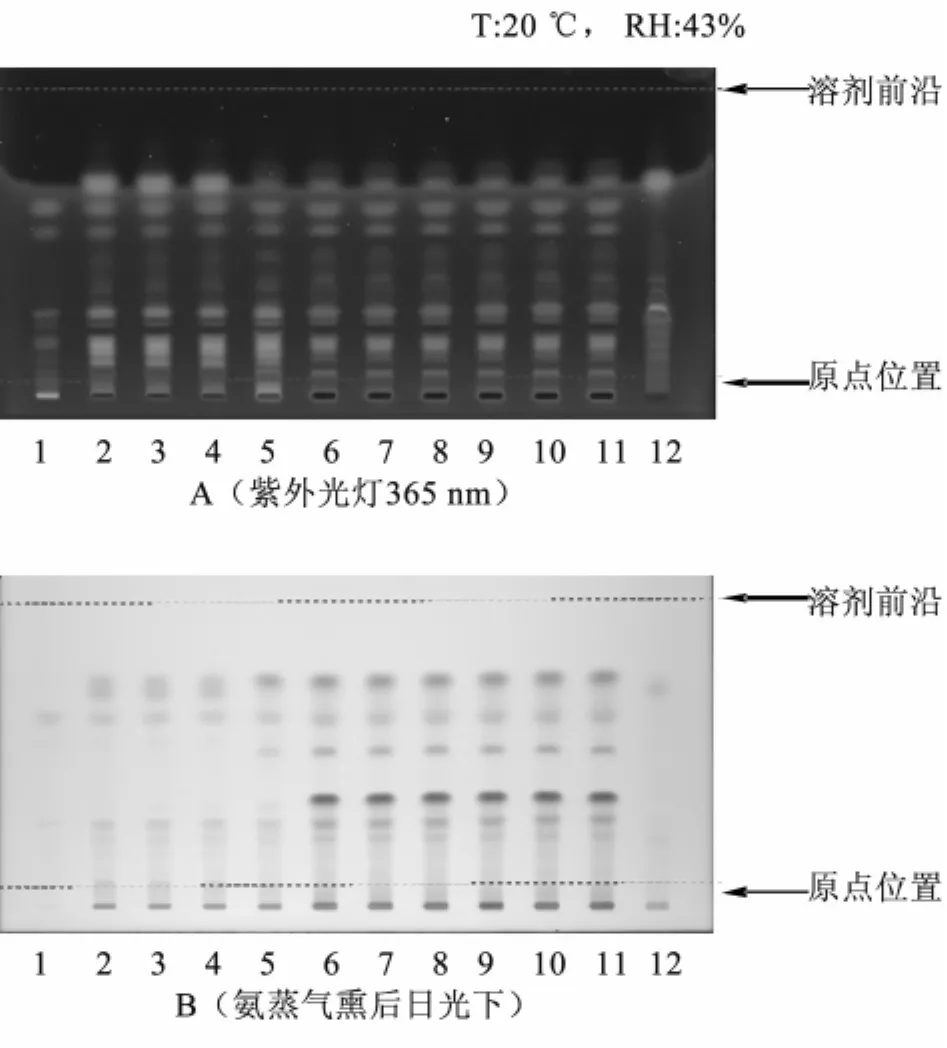
图1 紫草鉴别的薄层色谱图
2.2 黄芩的薄层色谱鉴别 黄芩所含的黄酮类主要化合物黄芩苷、黄芩素和汉黄芩素等均为多酚羟基化合物,显酸性,故采用紫草鉴别中的供试品溶液。取黄芩素对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。同时取处方中除去黄芩的其他药味,按处方比例制成缺黄芩的阴性对照样品。取阴性对照样品约20 g,同供试品溶液的制备方法制成阴性对照溶液。照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,吸取供试品溶液、对照药材溶液各5 μL,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲醇-甲酸(10∶3∶1∶2)为展开剂,展开,取出,晾干,喷以 5% 三氯化铁乙醇溶液,在105℃下加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性对照无干扰,见图2。
2.3 大黄的薄层色谱鉴别 大黄所含大黄素、大黄素甲醚和大黄酚等均为酚羟基蒽醌衍生物,显酸性,故也可采用紫草鉴别中的供试品溶液。取大黄素甲醚和大黄酚对照品,加乙酸乙酯制成每1 mL各含0.5mg的混合溶液,作为对照品溶液。同时取处方中除去大黄的其他药味,按处方比例制成缺大黄的阴性对照样品。取阴性对照样品约20 g,同供试品溶液的制备方法制成阴性对照溶液。照薄层色谱法(《中国药典》2010年版一部附录Ⅵ B)试验,吸取供试品溶液10 μL、对照品溶液2 μL,分别点于同一硅胶G薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。置氨蒸气中熏至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性对照无干扰,见图3。
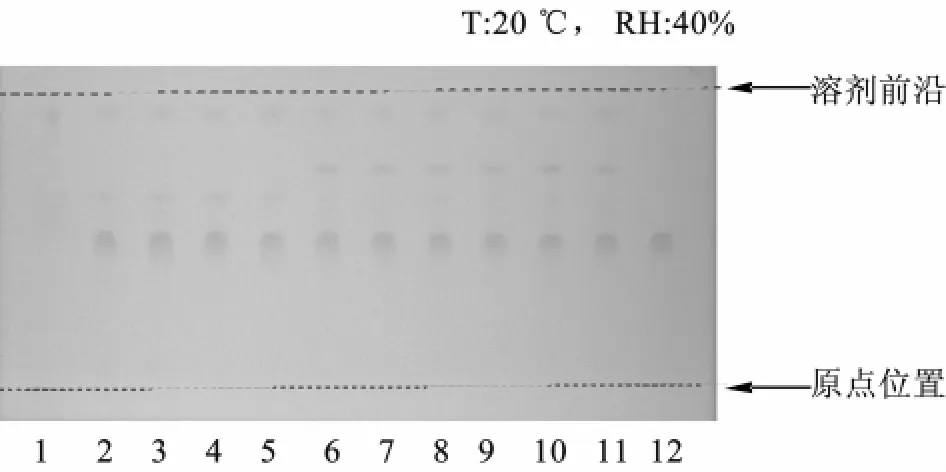
图2 黄芩鉴别的薄层色谱图
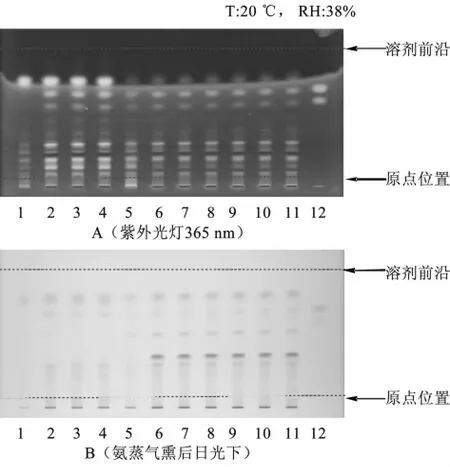
图3 大黄鉴别的薄层色谱图
3 含量测定
处方中的马尾连、大黄、紫草、地榆、黄芩均使用油炸提取,常用的指标成分大黄素、左旋紫草素和黄芩苷等已发生部分转化[3],因此选择处方中具有清热止痛作用的冰片为含量测定指标,用水杨酸甲酯为内标的毛细管气相色谱法进行测定。
3.1 色谱条件 弹性石英毛细管柱(柱长30 m,内径0.25 mm,膜厚度 0.25 μm)HP -FFAP;程序升温:初始温度 130℃,保持15 min,以每分钟30℃的速率升温至200℃,保持5 min;检测器为氢火焰离子化检测器(FID),检测器温度为300℃;进样口温度为210℃;恒压11.47 PSi;分流比10∶1;进样量 1 μL。
3.2 对照品溶液的制备 精密称取冰片对照品20.36 mg,加乙酸乙酯溶解并稀释至50 mL,作为对照品储备液(浓度为:0.407 2 mg·mL-1)。精密称取水杨酸甲酯 86.18 mg,加乙酸乙酯稀释至100 mL,作为内标溶液(浓度为:0.861 8 mg·mL-1)。精密量取对照品储备液5 mL,置10 mL量瓶中,精密加入内标溶液2 mL,加乙酸乙酯稀释至刻度,摇匀,即得。
3.3 供试品溶液的制备 精密称取本品0.5 g,置10 mL量瓶中,精密加入内标溶液2 mL,加乙酸乙酯稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。
3.4 线性范围 分别精密吸取3.2项下对照品储备液2 mL、4 mL、6 mL、8 mL、10 mL,置 10 mL 量瓶中,分别加乙酸乙酯稀释至刻度,摇匀,各精密吸取1 μL,注入气相色谱仪测定。以对照品的进样量为横坐标,异龙脑和龙脑的峰面积之和为纵坐标,进行线性回归,得回归方程Y=1 680.044 2 X+12.219 6,相关系数 r=0.999 5。结果表明,冰片进样量在0.081 44~0.407 2 μg之间与峰面积积分值呈良好的线性关系。
3.5 仪器精密度试验 取3.2项下的对照品溶液按确定的色谱条件,连续进样6次,以冰片中龙脑和异龙脑的总面积计算平均校正因子及RSD,计算公式:校正因子(AS:内标物峰面积;CS:内标物浓度;AR:对照品峰面积;CR:对照品浓度)结果平均校正因子 f值为0.684 6,RSD为0.29%。结果表明本方法精密性良好。
3.6 稳定性试验 取同一供试品溶液,每隔2 h进样一次,每次1 μL,共考察8 h,测定各异龙脑与龙脑的峰面积之和与水杨酸甲酯峰面积的比值,以观察样品溶液在检测过程中待测成分的稳定性。结果表明供试液在8 h内异龙脑与龙脑的峰面积之和与水杨酸甲酯峰面积的比值RSD为0.69%,供试品溶液在8 h内稳定性良好。
3.7 重复性试验 取烫伤油(广西 批号080304)按照供试品溶液的制备方法,平行制备6份样品,按上述方法和条件进行测定,计算每份供试品的含量。结果平均含量为3.2 mg·g-1,RSD为1.2%,说明本品含量测定方法的重复性良好。
3.8 加样回收率 精密称取已知含量为3.2 mg·g-1的样品(广西 批号080304)0.25 g,共6份,每份分别加入浓度为0.412 8 mg·mL-1的冰片对照品溶液2 mL,内标溶液2 mL,加乙酸乙酯使溶解并稀释至刻度,摇匀,滤过,吸取1 μL,注入气相色谱仪,分别计算加样回收率。结果见表1,表明测定方法的回收率良好。
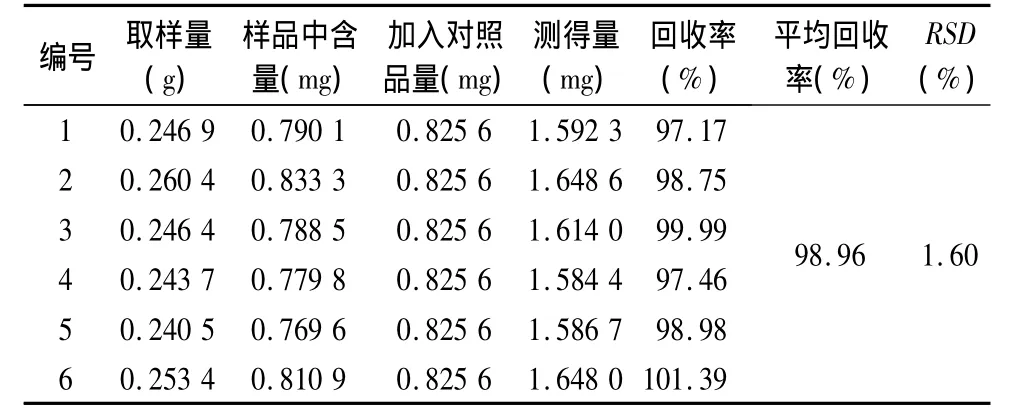
表1 加样回收率试验结果
3.9 空白试验 取处方中除去冰片的其他药味,按处方比例制成缺冰片的阴性对照样品。取阴性对照样品约0.5 g,同供试品溶液的制备方法制成阴性对照溶液。
分别精密吸取上述内标溶液、对照品溶液、供试品溶液及阴性对照溶液各1 μL,注入气相色谱仪,进行测定。结果供试品色谱中,在与对照品色谱和内标物质水杨酸甲酯保留时间相同的位置上,有相应色谱峰,而阴性对照色谱中无相应色谱峰。说明处方中其他药味对测定结果无干扰,见图4。
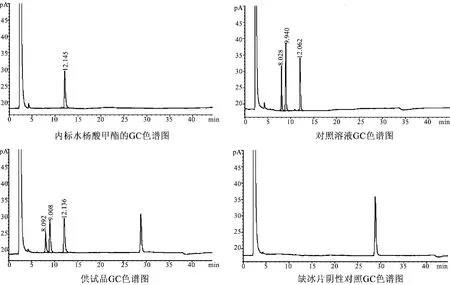
图4 空白试验GC色谱图
3.10 样品测定 按本文建立的方法分别测定了三家企业10批样品的含量(n=2),测定结果见表2。
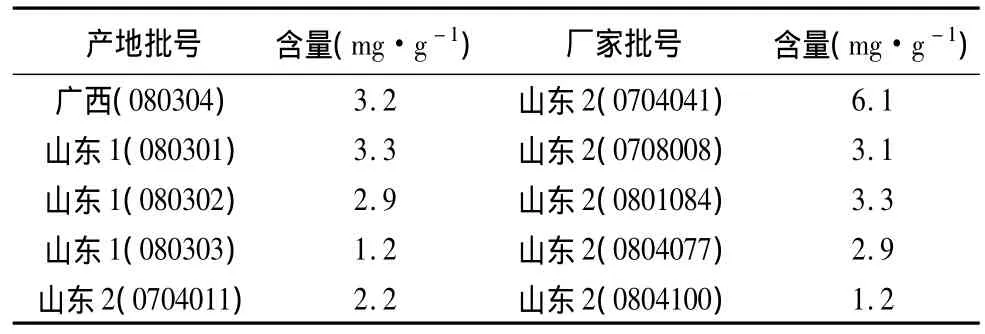
表2 10批样品中冰片含量测定结果
4 讨论
4.1 薄层鉴别制备供试品溶液时,曾采用乙醇萃取法[4]。但试验中发现乙醇萃取液浓缩后仍有大量麻油,无法进一步精制,提取特征成分。通过研究发现紫草、黄芩和大黄所含的特征成分均为多酚羟基化合物,具一定的酸性。因此采用酸碱水溶液处理方法既能很好地从麻油中提取特征成分,又能同时提取紫草、黄芩和大黄三味药材中的多酚羟基成分,实现一个供试品溶液供多味药材鉴别的目的,为同类剂型的质量分析提供参考。
4.2 紫草的薄层色谱鉴别方法建立时曾尝试以紫草对照药材和左旋紫草素对照品为对照进行试验,结果供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,未见相同颜色的斑点。由于制备工艺中经过油炸工序,紫草所含的萘醌类化合物可能转化为其他物质。故将紫草对照药材同样通过油炸后,提取制备对照药材溶液,结果供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。黄芩的薄层色谱鉴别方法曾尝试以黄芩苷、黄芩素和汉黄芩素为对照进行试验,结果供试品色谱中未检出黄芩苷,可能是黄芩苷遇高温被破坏,检出黄芩素和汉黄芩素,但汉黄芩素斑点不清晰,故最终选择黄芩素作对照。
4.3 本试验还对烫伤油的色谱条件进行了最优条件的选择,考察了不同的色谱柱:Agilent HP-FFAP、Agilent Innowax和Agilent DB-1701,结果三种色谱柱测定冰片,对称因子、分离度和理论板数等均符合要求[5],选择该色谱条件测定冰片,对色谱柱选择性不强,适用范围广。还比较了不同的柱温:110℃恒温测定,各成分峰都达到较好的分离,但由于保留时间较长,影响峰形;150℃恒温测定,异龙脑和龙脑的分离度达不到要求;130℃恒温测定,各成分峰都达到较好的分离,且峰形较好,但供试品色谱中,待测成分检出后,仍有杂质峰出现。为了避免杂质峰影响及节约分析时间,最终选择3.1项下的色谱条件。由表2测定结果可以看出,三家企业十批样品的含量从1.2到6.1 mg·g-1,差异达5倍。说明由于原质量标准未对冰片进行质量控制,企业产品批间均一性较差。因此,通过对烫伤油中冰片质量分析方法的建立,可有效控制产品的内在质量。
[1]山东省卫生厅.山东省药品标准[S].济南:山东省卫生厅,1986.
[2]中华人民共和国卫生部药典委员会.中华人民共和国卫生部药品标准中药成方制剂(第二册)[S].北京:人民卫生出版社,1990.
[3]叶勇,潘渭樵.温度对紫草素及其衍生物稳定性的影响[J].中国药物与临床,2007,7(7):540 -541.
[4]陈晶,马学礼,何艳萍.复方紫草油质量控制方法的研究[J].宁夏医学杂志2007,29(12):1147.
[5]国家药典委员会.中华人民共和国药典2010年版(一部)[S].北京:中国医药科技出版社,2010:附录38.
