HPLC-PAD法同时测定精制银翘解毒胶囊中对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷的含量
2012-09-20仇其原邓晓文姚立娟
仇其原,邓晓文,姚立娟
江苏省盐城药品检验所,盐城224001
HPLC-PAD法同时测定精制银翘解毒胶囊中对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷的含量
仇其原,邓晓文,姚立娟
江苏省盐城药品检验所,盐城224001
目的:采用HPLC-PAD法同时测定精制银翘解毒胶囊中对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷的含量。方法:色谱柱:Capcell Pak C18(150mm×4.6mm,5μm);流动相:乙腈-水(含0.25%的冰醋酸)梯度洗脱;检测波长:300 nm和228 nm;流速:1m L·m in-1;柱温:30℃。结果:对乙酰氨基酚测定的线性范围28.15~84.45μg(r=0.9999),平均含量为235.06mg·g-1,RSD为0.17%;绿原酸测定的线性范围0.2048~0.6144μg(r=0.9998),平均含量为1.91mg·g-1,RSD为0.21%;连翘苷测定的线性范围0.1054~0.3162μg(r=0.9994),平均含量为1.00mg·g-1,RSD为0.32%;牛蒡苷测定的线性范围1.044~3.132μg(r=0.9998),平均含量为7.04mg·g-1,RSD为0.16%。结论:该法简便、灵敏、准确,适用于精制银翘解毒胶囊的质量控制。
精制银翘解毒胶囊;对乙酰氨基酚;绿原酸;连翘苷;牛蒡苷;高效液相色谱法
精制银翘解毒胶囊处方由牛蒡子、金银花、甘草、连翘等10味中药和对乙酰氨基酚组成,具有清热散风、解毒退烧的功效。临床上用于治疗扁桃体炎、咽炎、上呼吸道感染等病症。精制银翘解毒胶囊目前使用的质量标准为国家标准[WS3-99(X-69)-99(Z)]。该标准含量测定项是用紫外分光光度法[1]仅对对乙酰氨基酚的含量进行了测定;该法操作繁琐,干扰因素多,容易引起误差。目前文献报道对乙酰氨基酚含量测定方法有TLCS法[2]、永停滴定法[3]、GC法[4]、HPLC法[5]等;而处方中同样具有重要疗效的中药如金银花的有效成分绿原酸、连翘的有效成分连翘苷、牛蒡子的有效成分牛蒡苷等均未做定量检测。
本文运用HPLC-PAD法[6]同时对对乙酰氨基酚[7]、绿原酸[8]、连翘苷和牛蒡苷4种成分含量进行检测;该法专属性高、重复性好、操作简便、结果准确;本法似可作为精制银翘解毒胶囊的含量测定方法,以更好地对该产品进行质量监控。
1 材料
1.1 仪器
Waters2695高效液相色谱仪、Waters2996二极管阵列紫外检测器(PAD)、Empower色谱工作站(美国waters公司);XS205 DU电子天平(瑞士梅特勒-托利多公司)。
1.2 试药
对照品:对乙酰氨基酚(批号100018~200408)、绿原酸(批号110753~200413)、连翘苷(批号110821~200711)、牛蒡苷(批号110819~201007)均购自中国药品生物制品检定所。
供试品:精制银翘解毒胶囊(贵州恒和制药有限公司,批号110205、111101);甲醇、乙腈(色谱纯);水(纯化水)。
2 方法与结果
2.1 色谱条件
2.1.1 不同流动相条件的考察吸取供试品溶液进样分析,分别以甲醇-水、乙腈-水、乙腈-0.4%磷酸、乙腈-0.1%磷酸、乙腈-0.5%冰醋酸、乙腈-0.25%冰醋酸[9]等不同浓度、不同比例的流动相系统进行等度和梯度洗脱实验,比较不同流动相系统以及不同梯度洗脱条件的色谱图,选择分离效果最好,时间尽可能短的最佳流动相条件。
2.1.2 检测波长的选择通过PAD检测器,对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷4种成分色谱峰于200~400 nm[10]波长扫描,选择合适波长进行实验。结果选择300 nm作为对乙酰氨基酚和绿原酸的检测波长;228 nm作为牛蒡苷和连翘苷的检测波长。
2.1.3 色谱条件最终选择的色谱条件:色谱柱为Capcell Pak C18(150mm×4.6mm,5μm);流动相为乙腈-水-冰醋酸,按表1进行梯度洗脱;检测波长为300 nm和228 nm;流速为1mL·min-1;柱温为30℃;进样量为20μL。理论塔板数按对乙酰氨基酚峰计不低于4000。

表1 乙腈-水(含冰醋酸0.25%)流动相梯度洗脱比例
2.2 溶液的配制
2.2.1 对照品溶液取对乙酰氨基酚28.15mg,置于10mL量瓶中,加50%甲醇溶解,定容至刻度,摇匀得每1mL含2.815mg对乙酰氨基酚的对照品溶液。另取绿原酸对照品10.24mg,置于100mL量瓶中,加50%甲醇溶解,定容至刻度,摇匀;另取2mL绿原酸溶液置10mL量瓶中,定容至刻度,制得每l mL含0.02048mg绿原酸的对照品溶液。另取连翘苷对照品10.54mg,置于100mL量瓶中,加50%甲醇溶解,定容至刻度,摇匀;另取5mL连翘苷溶液置50mL量瓶中,定容至刻度,制得每lmL含0.01054mg连翘苷的对照品溶液。另取牛蒡苷对照品10.44mg,置于100mL量瓶中,加50%甲醇溶解,定容至刻度,摇匀,制得每lmL含牛蒡苷0.1044 mg的对照品溶液。
2.2.2 供试品溶液取精制银翘解毒胶囊10粒,精密称定,内容物研细后,称取0.5956 g,置于50 mL的量瓶中,加50%的甲醇40mL,浸渍约30min,超声处理30min,放冷,加甲醇稀释至刻度,摇匀,滤过,取续滤液,用0.45μm微孔滤膜滤过,即得[11]。
2.2.3 空白对照溶液分别按缺少对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷的处方比例进行配制,方法见“2.2.2”项下,得到空白对照溶液。分别绘制两个波长的空白对照图谱,见图1。

图1 空白对照图谱(A)300 nm;(B)228 nm
2.3 系统适应性试验
按所给色谱条件将对照品溶液、阴性对照品、供试品溶液分别进样20μL,测定峰面积并记录色谱图,结果供试品色谱峰(见图2、图4)中,在各对照品色谱峰(见图3、图5)的相应位置上有保留时间相同的色谱峰;并且阴性样品(见图1)在与对照品色谱峰相应的位置上无对应的色谱峰出现,阴性样品对测定结果无干扰。对乙酰氨基酚峰、绿原酸峰、连翘苷峰和牛蒡苷峰的理论塔板数均高于4000,分离度大于2.0,符合要求。

图2 供试品在228 nm处色谱图峰3为连翘苷;峰4为牛蒡苷
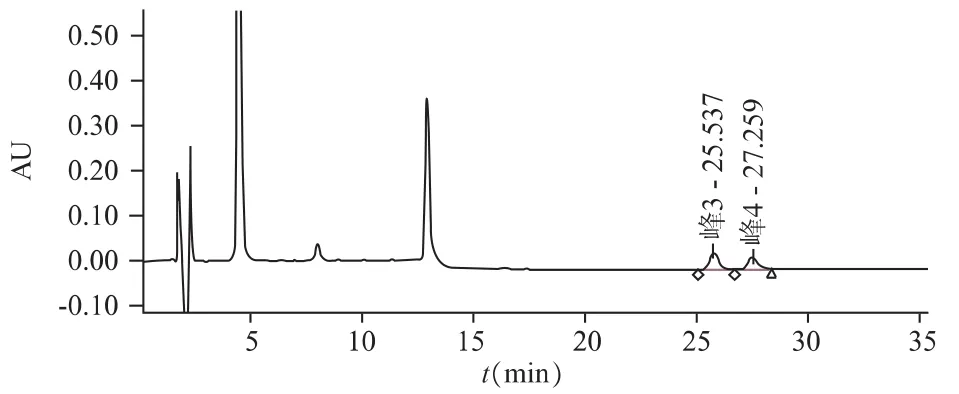
图3 对照品在228 nm处色谱图峰3为连翘苷;峰4为牛蒡苷

图4 供试品在300 nm处色谱图峰1为对乙酰氨基酚;峰2为绿原酸
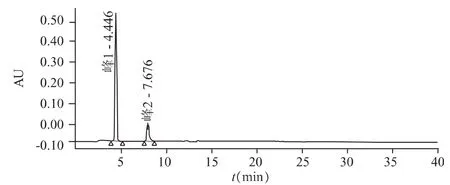
图5 对照品在300 nm处色谱图峰1为对乙酰氨基酚;峰2为绿原酸
2.4 线性关系的考察
取对照品储备液,按上述色谱条件分别进样10、15、20、25、30μL测定峰面积,以峰面积为纵坐标,进样量(μg)为横坐标,绘制标准曲线,对乙酰氨基酚的回归方程为:Y=1.492×105X-1.191×104,r= 0.9999,线性范围在28.15~84.45μg,呈良好线性关系;绿原酸的回归方程为:Y=2.179X×106-1.191× 104,r=0.9998,线性范围在0.2048~0.6144μg,呈良好线性关系;连翘苷的回归方程为:Y=5.085X×105-1.293×105,r=0.9994,线性范围在0.1054~0.3162μg,呈良好线性关系;牛蒡苷的回归方程为:Y=1.526X× 106-6.118×105,r=0.9998,线性范围在1.044~3.132 μg,呈良好线性关系。
2.5 精密度试验
取各对照品溶液,进样量为10μL,分别连续进样5次,测定峰面积,结果对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷峰面积的RSD分别为0.27%、0.21%、0.32%和0.36%。表明精确度良好。
2.6 重复性试验
取同一供试品溶液连续5次进样,测定该份样品中对乙酰氨基酚平均含量为235.06mg·g-1,RSD为0.68%;绿原酸平均含量为1.91mg·g-1,RSD为0.72%;连翘苷平均含量为1.00 mg·g-1,RSD为0.48%;牛蒡苷平均含量为7.04 mg·g-1,RSD为0.76%。表明本方法重复性良好。
2.7 稳定性试验
取样品(批号:111101)内容物1份,按照“2.2.2”项下配制供试品方法操作,按照“2.1”色谱条件分析,分别在0、4、12、24、48 h测定,结果5次所得的对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷峰面积值的RSD均小于2%,表明供试品溶液在48 h内稳定。
2.8 加样回收率试验
取样品(批号:111101)内容物9份[12],每份约0.28 g,精密称定,分别置于50mL量瓶中。由平均装量和样品测定项下所得含量(mg/粒)计算对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷的含量(mg),每份各加入一定量的对照品,加50%甲醇使溶解并稀释至刻度,经0.45μm微孔滤膜过滤,进样20μL,按外标法计算9份溶液中对乙酰氨基酚、绿原酸、连翘苷和牛蒡苷的量,结果见表2。
2.9 样品测定
取样品(批号:111101)20粒,称得平均装量,将内容物混匀,研细,称取约0.6 g,置于50mL量瓶中,加50%的甲醇40mL,浸渍约30min,超声处理30min,放冷,加甲醇稀释至刻度,摇匀,滤过,取滤液作为供试品溶液,进样20μL,按外标法以峰面积计算样品中对乙酰氨基酚、绿原酸、连翘苷及牛蒡苷的含量(mg/粒),同法测定另外一批的含量,结果见表3。
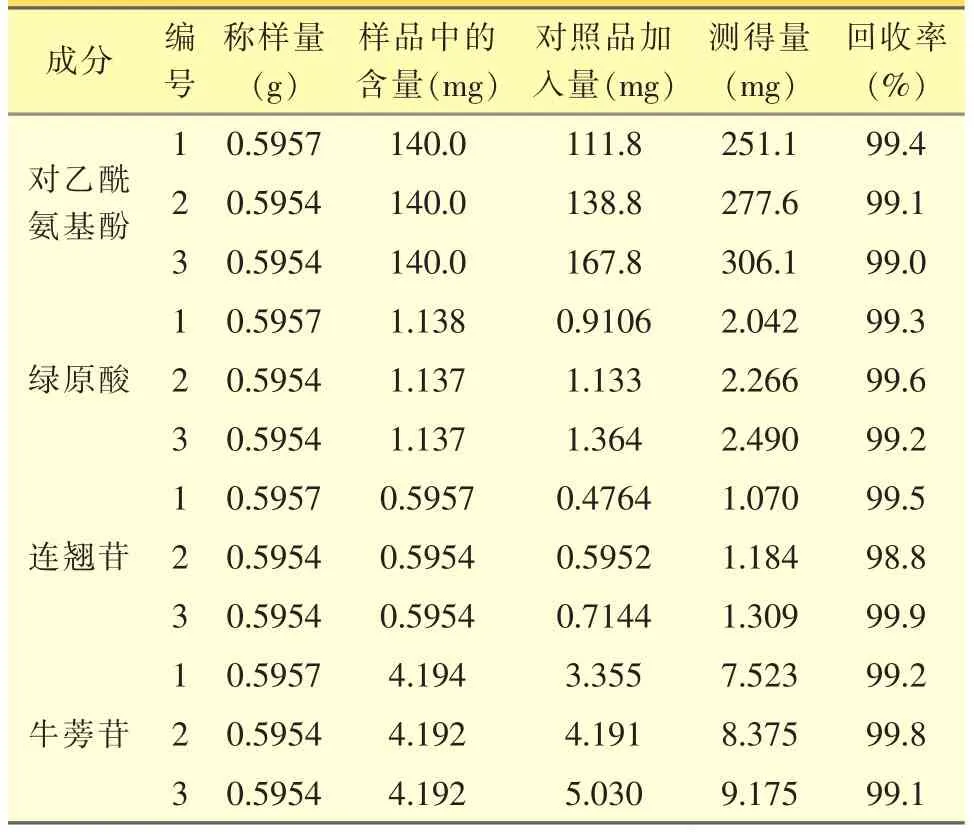
表2 各成分加样回收率试验
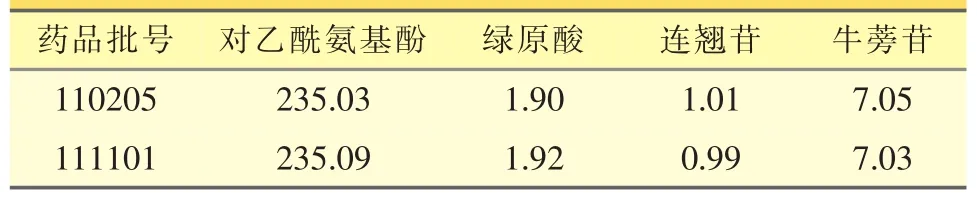
表3 样品含量测定结果(mg·g-1)
3 讨论
3.1 色谱条件的选择
3.1.1 波长的选择选择波长为228 nm和300 nm。通过各对照品溶液在200~400 nm处进行紫外光谱扫描检测显示,对乙酰氨基酚最大吸收波长为245 nm,绿原酸最大吸收波长为326 nm,连翘苷最大吸收波长为228 nm,牛蒡苷最大吸收波长为227 nm。在300 nm处,对乙酰氨基酚和绿原酸均有吸收,且线性关系良好,兼顾各组分的检测信号强度和检测灵敏度,故选择300 nm作为检测波长;牛蒡苷和连翘苷在228 nm附近处均有最大吸收,故选择228 nm作为检测波长。
3.1.2 流动相的考察精制银翘解毒胶囊组成复杂,其中各种活性成分的化学性质差异较大。本实验在设计过程中分别考察甲醇-水、乙腈-水、乙腈-0.4%磷酸、乙腈-0.1%磷酸、乙腈-0.5%冰醋酸、乙腈-0.25%冰醋酸等不同浓度、不同比例的流动相系统,进行等度和梯度洗脱实验,比较不同流动相系统以及不同梯度洗脱条件的色谱图,选择分离效果最好、时间尽可能短的最佳流动相条件。而采用乙腈-0.25%冰醋酸作流动相,进行梯度洗脱可将4个组分完全分离。
3.2 供试品溶液提取条件的考察
3.2.1 提取时间的优化在超声提取时,比较了15、30、45、60min含量测定结果,结果表明,提取时间30、45、60min时,各组分峰面积不随时间延长而有所增加,故超声处理时间选用30min。
3.2.2 提取溶剂的选择取供试品(批号:111101)研细后精密称取约0.6 g,分别以有机溶剂甲醇、50%甲醇、乙醇、50%乙醇为溶剂,对乙酰氨基酚等4种成分含量测定分析,实验表明,提取溶剂为50%甲醇时,提取效率最高,故选择其作为提取溶剂。
3.2.3 提取方式的选择取供试品(批号:111101)研细后精密称取约0.6 g,精密加入50%甲醇20 mL,称定重量,分别采用超声处理和热回流的方式提取60min,测定供试品中各组分含量。实验表明,采用超声处理与加热回流的结果一致,对实验无影响,而超声处理更加简单易行,故选择超声处理。
3.3 色谱柱的考察
分别采用Capcell Pak C18色谱柱(150mm×4.6 mm,5μm)、Diamonsil C18色谱柱(250mm×4.6mm,5μm)和Waters2695高效液相色谱仪,测定样品中对乙酰氨基酚等成分的含量,长柱分离效果好,适合多种成分中药分离,但出峰时间相对较长;短柱出峰时间短且主峰理论板数和分离度均符合要求,故选择Capcell Pak C18色谱柱(150mm×4.6mm,5 μm)作为色谱柱。
[1]国家药品标准·新药转正标准[S].第16~26册,30.
[2]屈爱桃,孙利民,温爱萍,等.薄层扫描法测定三九感冒灵冲剂中对乙酰氨基酚及咖啡因的含量[J].中国药房,2003,14(6):365-6.
[3]王巧荣,宋志超,冯明霞.永停滴定法测定复方对乙酰氨基酚片中对乙酰氨基酚的含量[J].中国新医药,2003,2(5):94-5.
[4]艾玉锁,宋小京.GC法测定复方感冒灵片中对乙酰氨基酚和咖啡因的含量[J].中国药事,2000,14(1):36-7.
[5]林娟,陈自平.HPLC法测定氨咖黄敏胶囊中对乙酰氨基酚的含量[J].安徽医药,2004,8(3):222-3.
[6]王守箐,王宏,李冠忠.HPLC-二极管阵列检测器测定银黄胶囊中绿原酸、黄芩苷含量[J].山东医药工业,2003,22(2):365.
[7]盖轲,郑阿利.高效液相色谱法同时测定氨咖黄敏胶囊中对乙酰氨基酚和咖啡因的含量[J].中国药房,200,17(6):456.
[8]刘祥兰,刘重芳,张英,等.金银花中绿原酸提取工艺的比较和优化研究[J].中成药,2000,22(6):402.
[9]林茂,朱朝德,孙庆民,等.当归化学成分的研究[J].药学学报,1979,14(9):529.
[10]付向红,付春华.RP-HPLC测定感冒灵胶囊中对乙酰氨基酚、绿原酸的含量[J].中国中医药信息杂志,2004,11(10):878-9.
[11]王学红,梁建平,郭志廷,等.贯叶连翘中金丝桃素的提取及含量测定方法[J].黑龙江畜牧兽医,2011,13(1):44-7.
[12]栾雨,曹云飞.RP-HPLC法测定抗感胶囊中绿原酸的含量[J].江苏大学学报(医学版),2006,16(1):64-5.
Simultaneous Quantitative Determ ination of Paracetamol,Chlorogenic Acid, Forsythin and Arctinin in Jingzhi Yinqiao Jiedu Capsules by HPLC-PAD
QIU Qi-yuan,DENG Xiao-wen,YAO Li-juan
Yancheng Institute of Pharmaceutical Control,Yancheng 224001,China
Objective:To establish an HPLC method for the determination of paracetamol,chlorogenic acid,forsythin and arctinin in Jingzhi Yinqiao Jiedu capsules.Methods:The column was Capcell Pak C18(150mm×4.6mm,5μm);Acetonitrile-water(It contained 0.25%glacial acetic acid)constituted the mobile phase for gradient elution;The wavelengths for detection were 300 nm and 228 nm;The flow rate was at 1 mL·min-1;The column temperature was at 30℃.Results:For paracetamol,the linear range was 28.15~84.45μg(r=0.9999),the average content was 235.06mg·g-1and the Relative Standard Deviation(RSD)was 0.17%;For chlorogenic acid,the linear range was 0.2048~0.6144μg(r=0.9998),the average content was 1.91mg·g-1and the RSD was 0.21%;For forsythin,the linear range was 0.1054~0.3162μg(r=0.9994),the average content was 1.00mg·g-1and the RSD was 0.32%;For arctinin,the linear range was 1.044~3.132 μg(r=0.9998),the average content was 7.04mg·g-1and the RSD was 0.16%.Conclusion:This method is accurate,simple and reproducible for quantitative analysis of Jingzhi Yinqiao Jiedu capsules.
Jingzhi Yinqiao Jiedu capsules;Paracetamol;Chlorogenic acid;Forsythin;Arctinin;HPLC
R927.2
A
1673-7806(2012)05-402-04
仇其原,男,主管药师E-mail:gita_80@126.com
2012-06-15
2012-07-25
