璇机化积膏质量标准研究
2012-09-06常增荣傅欣彤
李 姣, 常增荣, 傅欣彤
(1.北京市药品检验所,北京 100035;2.北京中医药大学中药学院,北京 100102)
璇机化积膏为国际传统医药研究院研制的贴膏剂,由香附、木香、五灵脂、牵牛子等药味与适宜的基质和基材制成。为控制药品的内在质量,对处方中木香、香附、牵牛子进行了薄层色谱鉴别,并采用高效液相色谱法测定了制剂中α-香附酮。所建立的方法具有分离效果好、重复性好,专属性强,准确等优点,可以有效地控制本品的质量。
1 仪器与试药
1.1 仪器 岛津LC-20A高效液相色谱仪,配备二极管阵列检测器 (PDA)型;硅胶G预制板 (烟台市化学工业研究所);硅胶G预制板 (青岛海洋化工厂分厂);MN-硅胶G预制板TLC-plate SIL G-25。
1.2 对照品及对照药材 去氢木香内酯对照品 (批号1525-200103)、α-香附酮对照品 (批号110748-200507)、咖啡酸对照品 (批号110885-200102)。香附对照药材 (批号121059-200505)、牵牛子对照药材 (批号121024-200604),均购于中国药品生物制品检定所。璇机化积膏样品3批(批号分别为20100616,20100621,20100816),由国际传统医药研究院提供;甲醇、乙腈为色谱纯,水为超纯水,其他试剂均为分析纯。
2 薄层色谱鉴别
2.1 木香中去氢木香内酯的薄层鉴别 取本品1片,除去盖衬,剪成小块,加甲醇30 mL,加热回流30 min,放冷,滤过,滤液加中性氧化铝5 g,充分振摇1 min,滤过,滤液浓缩至1 mL,取上清液,作为供试品溶液。另取不含木香的阴性样品制剂,同法制成木香阴性样品溶液。再取去氢木香内酯对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。照薄层色谱法试验,吸取上述3种溶液各5 μL,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯(20∶1)为展开剂,展距为12 cm,喷显色剂5%香草醛硫酸溶液后,热风加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。缺木香阴性样品无相应斑点。见图1。

图1 璇机化积膏中木香的TLC图谱
2.2 香附的薄层色谱鉴别 取本品3贴,除去盖衬,剪碎,置250 mL圆底烧瓶中,加水100 mL,连接挥发油测定器,自测定器上端加水使充满刻度部分并溢流入烧瓶时为止,再加乙酸乙酯1 mL,加热回流至沸,并保持微沸2 h,放冷,分取乙酸乙酯液,作为供试品溶液。取不含香附的空白制剂,同法制成阴性样品溶液。取香附对照药材1 g,置250 mL圆底烧瓶中,同法制成对照药材溶液。再取α-香附酮对照品,加乙酸乙酯制成每1 mL含1 mg的溶液,作为对照品溶液。照薄层色谱法试验,吸取上述4种溶液各2 μL,分别点于同一硅胶GF254薄层板上,以正己烷-乙酸乙酯(8.5∶1.5)为展开剂,展开,取出,晾干,置紫外光灯 (254 nm)下检视观察。供试品色谱中,在与对照药材及对照品色谱相应的位置上,分别显相同颜色的斑点。缺香附阴性样品无相应斑点。见图2。

图2 璇机化积膏中香附的TLC图谱
2.3 牵牛子的薄层色谱鉴别 取本品3贴,除去盖衬,剪碎,置烧杯中,加水70 mL,煎煮20 min,放冷,滤过,滤液加盐酸调节至pH值为1~2,用乙酸乙酯30 mL振摇提取,分取乙酸乙酯液,蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。取不含牵牛子的阴性样品制剂,同法制成阴性样品溶液。另取牵牛子对照药材2 g,加水70 mL,煎煮20 min,放冷,离心,取上清液,加盐酸调节至pH值为1~2,用乙酸乙酯30 mL振摇提取,分取乙酸乙酯液,蒸干,残渣加甲醇1 mL使溶解,作为对照药材溶液。再取咖啡酸对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。照薄层色谱法试验,吸取上述4种溶液各5 μL,点于同一高效硅胶G薄层板上,以二氯甲烷-甲醇-甲酸(93∶9∶4)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视观察。供试品色谱中,在与对照药材和对照品色谱相应的位置上,分别显相同颜色的荧光斑点。缺牵牛子阴性样品无相应斑点。见图3。

图3 璇机化积膏中牵牛子的TLC图谱
3 香附中α-香附酮的HPLC法测定
3.1 色谱条件 色谱柱为Phenomenex Gemini 5μm110ÅC18(4.6 mm×250 mm);流动相为甲醇-乙腈-水 (55∶5∶40);柱温为40℃,检测波长为254 nm。理论板数按α-香附酮峰计算应不低于6 000。
3.2 对照品溶液的制备 取α-香附酮对照品适量,精密称定,加甲醇制成每1 mL含5 μg的溶液,即得。
3.3 供试品溶液的制备 取本品5片,除去盖衬,剪除不含药部分的底衬,将含药部分对折 (以防混匀及取样过程黏连),称定总质量,剪碎,混匀,取约1片的量,精密称定,置具塞锥形瓶中,加乙酸乙酯20 mL,加热回流30 min,取出,加甲醇50 mL,振摇均匀,置水浴上继续回流10 min,放冷,滤过,滤液置100 mL量瓶中,用甲醇分次洗涤药渣及滤器,洗液并入量瓶中,加甲醇稀释至刻度,摇匀,取10 mL,置具塞锥形瓶中,加碱性氧化铝 (100~200目)3 g,振摇2 min,置冷水浴中放置15 min后,取上清液,用微孔滤膜 (0.45 μm)滤过,取续滤液,即得。
3.4 阴性样品溶液的制备 按处方比例及制剂制备工艺制备不含香附的阴性样品,再按3.3项下方法制备,即得。
3.5 专属性试验 取对照品溶液、供试品溶液和阴性样品溶液各10 μL,分别注入液相色谱仪,依法测定,结果阴性样品在α-香附酮相应保留时间处未见色谱峰,表明阴性样品对检测无干扰。见图4。
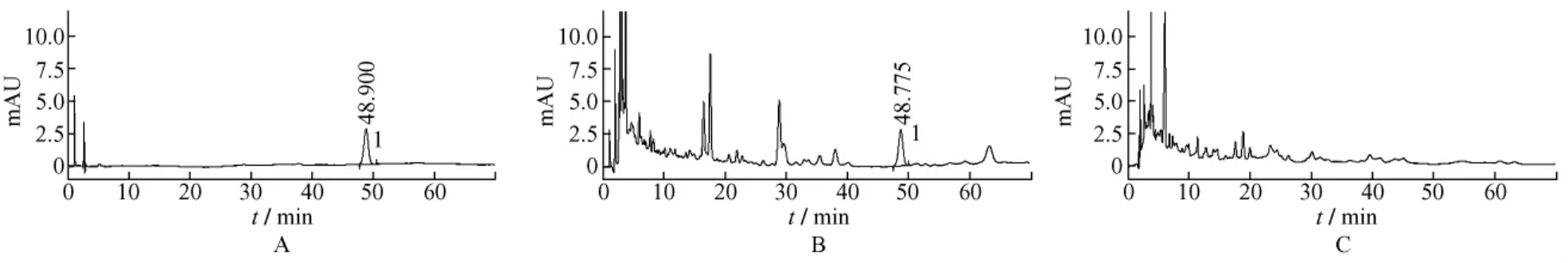
图4 α-香附酮测定HPLC图谱
3.6 线性关系考察 分别精密吸取α-香附酮对照品溶液(质量浓度为 1.098 mg/mL)0.1、0.2、0.4、0.8、1.6、2.0、4.0 mL,分别置100 mL量瓶中,各加甲醇稀释至刻度,摇匀,分别精密吸取10 μL,注入高效液相色谱仪,记录依法测定峰面积,以α-香附酮对照品进样量为横坐标,峰面积为纵坐标,绘制标准曲线,其回归方程为:Y=2 576 661X+460(r=1.000 0),表明α-香附酮对照品在0.010 98 μg ~0.439 2 μg范围内线性关系良好。
3.7 精密度试验 取同一供试品溶液 (批号20100616),连续进样6次,每次10 μL,注入液相色谱仪,测定,结果α-香附酮峰面积RSD为0.96%。
3.8 稳定性试验 取同一份供试品溶液 (批号20100616),按定量测定方法,分别在0、2、4、6、11、19、28 h进样测定。结果α-香附酮峰面积RSD为1.09%,表明供试品溶液在配制后28 h内基本稳定。
3.9 重复性试验 取本品20片,除去盖衬,称定质量,剪碎,取约1片的质量 (共取6份),精密称定,依法测定。结果α-香附酮含量重复性良好,RSD为2.65%,方法重复性良好。
3.10 加样回收率试验 取约半片的量 (共取6份),精密称定,置具塞锥形瓶中,精密加入用乙酸乙酯配制的质量浓度为0.230 6mg/mL的α-香附酮对照品溶液1.0mL,再加入乙酸乙酯19 mL,按供试品溶液同法制备,测定。结果α-香附酮对照品平均回收率为99.02%,RSD为2.88%,见表1。
3.11 样品测定按上述测定方法,对测定3批璇机化积膏样品,结果见表2。
4 讨论
4.1 木香的薄层鉴别[1-3]中,贴剂用甲醇回流提取后,溶液中有部分脱敏胶辅料,致使点样困难,加中性氧化铝可吸附脱敏胶及一些其他杂质。木香中主要含有倍半萜内酯类成分去氢木香内酯和木香烃内酯,木香烃内酯对热不稳定,在本制剂中曾同时对去氢木香内酯和木香烃内酯进行检测,结果供试品在木香烃内酯相应位置上未检出清晰的薄层斑点,故仅以去氢木香内酯为对照。

表1 回收率试验结果

表2 样品中α-香附酮测定结果(n=3)
4.2 木香的薄层鉴别中曾对展距进行考察,结果展距为12 cm左右时,分离效果最好,故确定展距为12 cm,另对本薄层鉴别的耐用性进行了考察,考察项目为3个不同厂家的薄层板、不同展开温度 (室温25℃和低温6℃)、湿度 (相对湿度45%和75%),结果表明不同厂家薄层板、不同的温湿度条件下本薄层鉴别均能获得较好的分离效果。
4.3 贴膏剂的质量控制研究[4-10]中,最大问题是检测成分的提取和净化,本品为含亲脂性基质的贴剂,有较强的黏附性,为有效提取α-香附酮及尽可能少的带入基质,对提取溶剂进行预筛选,分别采用甲醇、乙醇、异丙醇、乙醚、乙酸乙酯、三氯甲烷提取,结果乙醚、三氯甲烷、乙酸乙酯能较好溶解药膏,但同时贴膏的赋形剂也较多溶出,用甲醇为溶剂提取液颜色较浅,表明甲醇不能较好渗透进本贴剂中,提取效果较差,同时,由于α-香附酮具有挥发性,供试品不宜采用蒸发等浓缩过程,故采用乙酸乙酯提取后加甲醇使赋形剂沉淀析出的提取方法。为进一步除去残留的赋形剂及制剂中其他黄酮等成分的干扰,又采用碱性氧化铝处理净化样品。
4.4 参照文献的方法[11-13],α-香附酮定量测定的流动相曾采用①甲醇-水 (75∶25),②甲醇-水 (70∶30),③甲醇-水 (65∶35),④甲醇-水 (60∶40),⑤乙腈-水 (60∶40),⑥乙腈-水 (55∶45),⑦乙腈-水 (45∶55)体积流量为1.0 mL/min,⑧甲醇-乙腈-水 (55∶5∶40),体积流量为1.0,柱温分别采用30℃与40℃,结果流动相⑧甲醇-乙腈-水 (55∶5∶40),柱温40℃,分离效果较好,故采用甲醇-乙腈-水 (55∶5∶40)为流动相,柱温40℃。
4.5 采用本文色谱条件用归一化法计算,α-香附酮对照品纯度为96.08%。
4.6 分别采用不同品牌色谱柱 (1)Phenomenex Gemini 5μm110ÅC18(4.6 mm ×250 mm),(2)COSMOSIL Packed Column 5C18-MS-Ⅱ (5μm,250 mm ×4.6 mm), (3)YMC Packed ProC18(5 μm,250 mm ×4.6 mm), (4)Inertsil ODS-3(5 μm,250 mm×4.6 mm),进行测定,结果α-香附酮峰在各种色谱柱均分离良好,空白无干扰,不同色谱柱间测定α-香附酮的RSD为2.42%,表明本HPLC方法有较好的耐用性。
[1]周 军,张 晶,金兆祥,等.济坤丸质量标准研究[J].天津药学,2010,22(1):28-30.
[2]李冬梅,刘永利.中国药典品种参苏丸中木香鉴别的商榷[J]. 时珍国医国药,2010,21(7):1820.
[3]姚令文,郑笑为,刘 燕,等.治痢片的质量标准研究[J].中国药品标准,2009,10(5):342-346.
[4]郭汉文,王怡君,孔黎冰,等.HPLC法测定骨友灵贴膏中延胡索乙素的含量[J].中国药品标准,2007,8(4):32-34.
[5]韩建伟,刘 颖,魏元锋,等.HPLC法测定痛经膏贴中黄芩苷和汉黄芩素含量研究[J].中国实验方剂学杂志,2008,14(8):8-10.
[6]郭 玫,余晓晖,陈卫林,等.咳喘灵贴膏质量标准研究[J].中国中医药信息杂志,2006,13(3):47-48.
[7]樊敏伟.如意金黄贴膏剂质量标准的研究[J].中成药,2005,27(7):864-866.
[8]刘建芳,毕开顺.肾泰贴膏中蛇床子素的含量测定[J].中国中药杂志,2002,27(10):745-746.
[9]刘卓妍,梁勇坤,徐履琴.胃痛贴膏的质量标准研究[J].中成药,2000,22(6):407-409.
[10]叶立新,张树平,赖素萍,等.乳癖宁贴膏中淫羊藿苷的含量测定方法[J].中国中药杂志,2005,30(12):944-945.
[11]许世辉.高效液相色谱法测定妇科调经片中α-香附酮的含量[J].海峡药学,2010,22(9):62-63.
[12]侯立静,吴丽丽,李英霞.不同产地香附和醋炙香附中α-香附酮含量测定[J].陕西中医,2011,32(4):480-481.
[13]翟志光,牛 欣,冯前进,等.HPLC测定良附口服液中α-香附酮的含量[J].中国中药杂志,2007,32(10):988-989.
