基于psbA-trnH分析的何首乌野生居群遗传多样性
2012-09-06白明明孙小芹郭建林李密密杭悦宇
白明明,孙小芹,郭建林,李密密,杭悦宇
〔江苏省·中国科学院植物研究所(南京中山植物园)江苏省植物迁地保护重点实验室,江苏 南京 210014〕
基于psbA-trnH分析的何首乌野生居群遗传多样性
白明明,孙小芹,郭建林,李密密,杭悦宇①
〔江苏省·中国科学院植物研究所(南京中山植物园)江苏省植物迁地保护重点实验室,江苏 南京 210014〕
对产自不同省区的何首乌〔Fallopia multiflora(Thunb.)Harald.〕17个野生居群85个单株的psbA-trnH序列进行了扩增和分析,在此基础上分析了居群间的遗传多样性,并采用NJ法对85个单株进行了聚类分析。结果表明:供试85个单株的psbA-trnH序列长度为384 bp;其中,变异位点为167 bp,简约信息位点为53 bp,分别占序列总长度的43.5%和13.8%。变异类型主要为碱基缺失和替换;变异位点主要集中在235~281 bp区域,根据位点变异情况可将17个居群大体分为3类。各居群间的遗传距离为0.000~0.172,其中,贵州居群与其他16个居群间的遗传距离为0.167~0.172,而其他16个居群间的遗传距离为0.000~0.017。17个居群间的核苷酸多样性指数(Pi)、遗传分化系数(Nst)和基因流(Nm)分别为0.028 56、0.918 68和0.04;除贵州居群外其他16个居群的Pi、Nst和Nm分别为0.015 68、0.837 19和0.10;贵州居群与其周边省区(四川、云南、广西、湖南和湖北)居群的Pi、Nst和Nm分别为0.047 99、0.937 62和0.03。在NJ系统树上,17个居群可聚为4支,且大部分居群的供试单株聚在同一分支中;仅贵州居群单独聚为一支,与序列分析的划分结果基本一致。由研究结果可见:野生何首乌居群总遗传变异的91.868%来自居群间,8.132%来自居群内,居群间的基因交流较少;除贵州居群外其余16个居群的整体遗传多样性水平偏低,说明何首乌居群整体多样性水平在很大程度上受贵州居群的影响。
何首乌;居群;psbA-trnH序列;遗传多样性;NJ系统树
何首乌〔Fallopia multiflora(Thunb.)Harald.〕为蓼科(Polygonaceae)何首乌属(FallopiaAdans.)多年生草本植物[1],其块根和藤茎均为传统中药材,具有解毒、消痈、截疟、润肠通便的功效[2]。何首乌野生资源丰富,主要分布在安徽、福建、广东、甘肃、广西、贵州、湖北、河南、湖南、江苏、江西、四川、山东、上海、陕西、云南和浙江。
目前,有关何首乌遗传多样性的研究非常少。王凌晖等[3]采用RAPD技术对广西产10个野生何首乌居群进行了遗传多样性分析,用40对随机引物共扩增出175个多态性片段,多态性百分率达59.3%;严寒静等[4]采用PCR直接测序法测定何首乌10个种源核糖体DNA的ITS序列,共找到22个变异位点,其中ITS1区域有4个、ITS2区域有16个、5.8S区域有2个,序列间遗传分化距离0.001 75~0.049 45。对于野生何首乌来说,上述研究涉及的样品分布范围并不广、居群数也偏少,且所用片段的变异位点不能充分反映何首乌野生种质资源的遗传结构。
psbA-trnH基因间隔区位于编码光合系统Ⅱ反应中心DⅠ蛋白的psbA基因和编码tRNA组氨酸的trnH基因之间,被认为是叶绿体基因组中进化速率最快的基因间隔区之一。目前,已将psbA-trnH序列分析用于薯蓣属(DioscoreaL.)3个种[5]、石斛属(DendrobiumSw.)15个种[6]和连翘〔Forsythia suspensa(Thunb.)Vahl〕3个居群[7]的遗传多样性研究。
作者对采自17个省区的野生何首乌85个单株的psbA-trnH序列进行了分析,比较分析野生何首乌的遗传多样性,探讨不同居群的遗传变异程度,以期为其优良品种的选育和种质资源的研究提供依据。
1 材料和方法
1.1 材料
供试的17个野生何首乌居群的分布地及基本地理信息见表1。凭证标本存放于江苏省·中国科学院植物研究所标本馆(NAS),经江苏省·中国科学院植物研究所杭悦宇研究员鉴定。于2010年在每个居群选取5株样株,采集幼嫩且无病虫害的干净叶片,用变色硅胶干燥后保存、待用。
实验用10×PCR buffer(含Mg2+)、TaqDNA聚合酶和dNTPs均购自上海博彩生物科技有限公司,引物由上海英骏生物技术有限公司合成,AxyPrep DNA凝胶回收试剂盒购自杭州爱思进生物技术有限公司。
1.2 方法
1.2.1 基因组总DNA的提取参照文献[8]采用改良CTAB法提取基因组总DNA。取10 mg叶片,在液氮中研磨后加入500 μL提取液于65℃水浴中保温45 min,期间摇动30次;加入500 μL体积比24∶1的氯仿-异戊醇混合液,混合均匀,12 000 r·min-1离心10 min;取上清液,加入2倍体积无水乙醇,-20℃放置1 h以上;4 000 r·min-1离心10~20 min使DNA沉淀;用700 μL体积分数70%乙醇清洗1次,4 000 r ·min-1离心5~10 min;再用700 μL无水乙醇清洗1次,4 000 r·min-1离心5~10 min;得到DNA沉淀,晾干,最后加入100 μL水或TE溶解DNA。
1.2.2psbA-trnH序列的扩增及产物检测根据GenBank中发布的何首乌psbA-trnH序列(GenBank登录号为EU554047.1),采用Primer Premier 5.0软件自行设计psbA-trnH序列引物,FHS和FHA引物序列分别为5'-TTCCCGCTAGACCTAGCTGC-3'和5'-ACTGCCTTGATCCACTTGGC-3'。
扩增反应在PE-9600型PCR仪(Perkin Elmer公司生产)上进行。扩增反应体系总体积20 μL,含30 ng模板DNA、10×PCR buffer(含Mg2+)2 μL、10 mmol·L-1dNTPs 0.3 μL、5 μmol·L-1引物2 μL和5 U·μL-1TaqDNA聚合酶0.2 μL,用灭菌双蒸水补足至20 μL。扩增程序为:94℃预变性5 min; 94℃变性40 s,58℃退火40 s,70℃延伸45 s,共30个循环;最后于72℃延伸5 min。

表1 供试何首乌野生居群的基本概况1)Table 1 Basic status of wild populations of Fallopia multiflora(Thunb.)Harald.tested1)
扩增产物与6×甘油凝胶上样液混合后,用质量体积分数0.8%琼脂糖凝胶(含0.5 μg·mL-11×EB)进行电泳检测,电泳时间约40 min。电泳结束后用WV-BP330型凝胶扫描分析系统(江苏捷达科技发展有限公司生产)观察扩增结果并拍照。
1.2.3 扩增片段的纯化和序列测定PCR扩增产物用AxyPrep DNA凝胶回收试剂盒进行纯化,纯化后使用引物FHS和FHA直接进行双向测序,测序由上海华大基因有限公司完成。
1.3 序列分析
采用Sequencher软件进行峰图文件的编辑和拼接,拼接好的序列用MEGA 4.1软件进行多重序列比对(multiple sequence alignment,MSA),对序列长度、保守位点、变异位点和简约信息位点等序列特征进行分析[9];通过Kimura 2-parameter(K2P)模型计算遗传距离,构建距离矩阵[10];应用DnaSP 5.10[11]软件计算核苷酸多样性指数[12]、基因流[13]和居群间遗传分化系数[13];以萹蓄(PolygonumaviculareL.) (GenBank登录号为FJ395458.1)为外类群,通过邻接法(neighbor-joining method,NJ)构建基于遗传距离的聚类图,构建的系统发育树采用1 000次循环的自举检验法(bootstrap test)检验各分支的置信度,空缺(gap)始终作为缺失(missing)处理[14]。
2 结果和分析
2.1 何首乌居群psbA-trnH序列扩增结果分析
供试的85个野生何首乌单株psbA-trnH序列的长度为243~434 bp,排序后两端切平获得的序列长度为384 bp,当空缺(gap)始终作缺失(missing)处理时,保守位点有217 bp,占序列总长度的56.5%;变异位点有167 bp,占序列总长度的43.5%,变异类型主要为碱基缺失和替换。其中,简约信息位点有53 bp,占序列总长度的13.8%。
2.2 何首乌居群psbA-trnH序列的差异分析
何首乌不同居群psbA-trnH序列变异位点的比较结果见表2。由表2可见:何首乌不同居群间和单株间psbA-trnH序列均存在碱基变异位点,主要集中在235~281 bp区域。福建、广东、湖北、湖南、江西、四川和浙江居群的所有单株,山东居群的4号单株,上海居群的2号和4号单株,广西居群的1~4号单株以及云南居群的2~5号单株的psbA-trnH序列在235~281 bp位点的碱基序列依次为TATGGCCTCT TTGGTGT(或G)TTTG(或T或A)GTTGGGTGGACT TTTTTTTGATTCAT;贵州居群5个单株的psbA-trnH序列在235~281 bp位点的碱基序列依次为TCTCCCTCTTT--TGTTTT----GGG-GGAAATTTCTTT--TTCTT(“-”表示碱基缺失)。比较而言,贵州居群psbA-trnH序列235~281 bp区域间有16个变异位点,与其他11个居群的差异较大。在251 bp位置,其他11个居群的碱基分别为T和G,其中,福建、湖北、江西、四川和浙江居群的所有单株,山东居群的4号单株,上海居群的2号和4号单株及云南居群的2~5号单株的碱基为T;广东和湖南居群的所有单株以及广西居群的1~4号单株的碱基为G。在255 bp位置,福建、湖北、江西和浙江居群的所有单株,山东居群的4号单株以及上海居群的2号和4号单株的碱基为G;广东和湖南居群的所有单株以及广西居群的1~4号单株的碱基为T;四川居群的5个单株和云南居群的2~5号单株的碱基为A。
安徽、甘肃、河南、江苏和陕西居群所有单株的psbA-trnH序列在235~281 bp位点的碱基均缺失;山东居群的1~3号和5号单株,上海居群的1号、3号和5号单株,广西居群的5号单株及云南居群的1号单株在这段序列位点也存在碱基缺失。
根据psbA-trnH序列235~281 bp区域的碱基差异,17个野生何首乌居群可被划分为3大组:第1组包括福建、广东、湖北、湖南、江西、四川、浙江、广西和云南居群;第2组仅贵州居群,其特征为235~281 bp序列与第1组相比有16个变异位点;第3组包括安徽、甘肃、河南、江苏、陕西、山东和上海居群,其特征为235~281 bp区域碱基缺失。另外,第1组又可分为3小组:第1小组包括广东、湖南和广西居群,其251 bp位点的碱基为G、255 bp位点的碱基为T;第2小组包括福建、湖北、江西和浙江居群,其251和255 bp 2个位点的碱基恰好与第1小组完全相反;第3小组包括四川和云南居群,其251和255 bp的碱基分别为T和A。
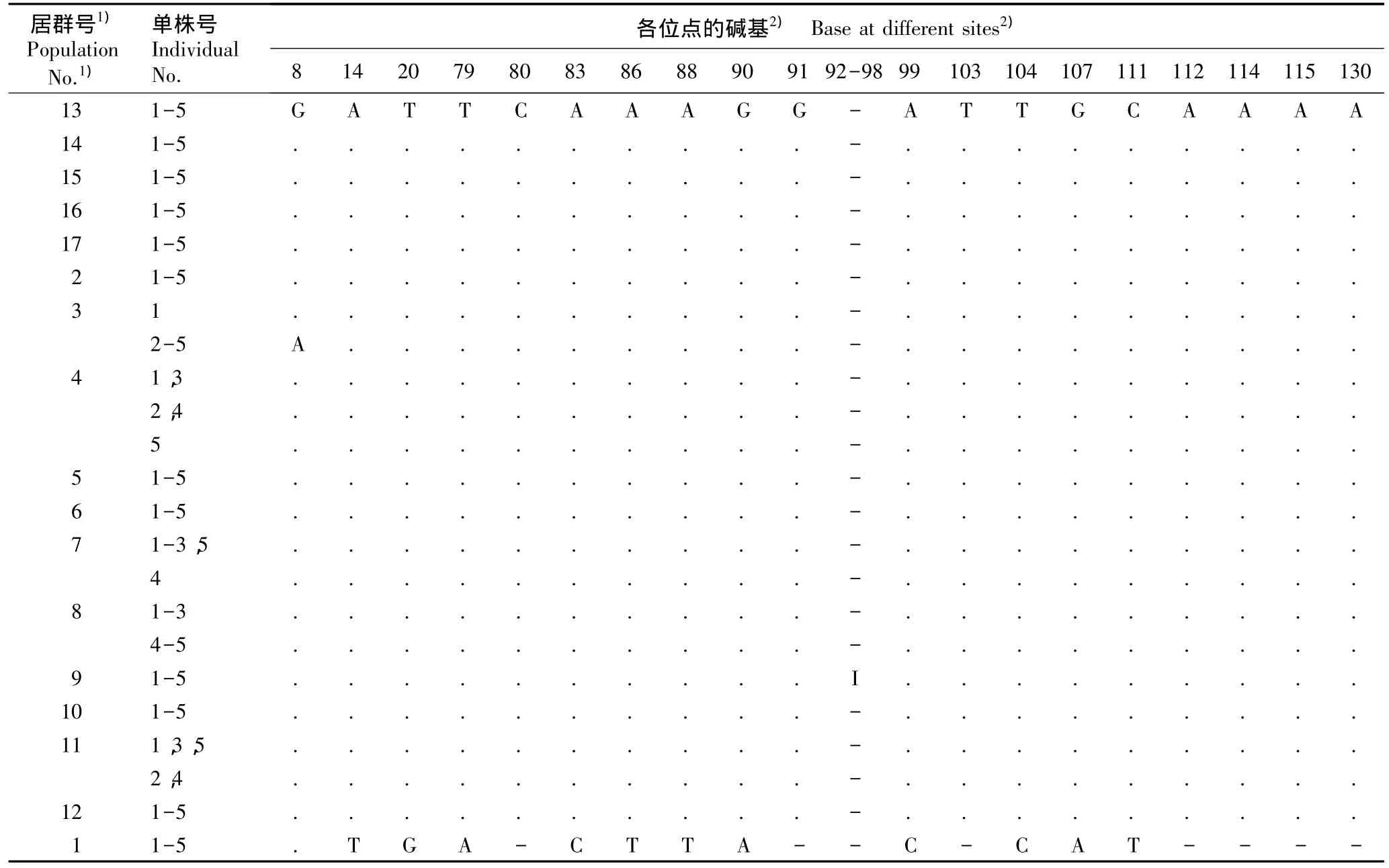
表2 何首乌17个野生居群psbA-trnH序列变异位点的比较Table 2 Comparison of variable sites in psbA-trnH sequence of seventeen wild populations of Fallopia multiflora(Thunb.)Harald.

续表2Table 2 (Continued)

续表2>Table 2 (Continued)
2.3 何首乌居群的遗传多样性分析
采用MEGA 4.1软件中的Kimura 2-parameter模型计算何首乌不同居群间psbA-trnH序列的遗传距离并构建序列距离矩阵,结果见表3。何首乌各居群间的遗传距离为0.000~0.172;其中,贵州居群与其他居群间的遗传距离均较大,为0.167~0.172;其他居群间的遗传距离均较小,为0.000~0.017。
何首乌17个野生居群的核苷酸多样性指数为0.028 56±0.005 43,居群间的遗传分化系数达到0.918 68,基因流为0.04。说明17个何首乌野生居群总遗传变异的91.868%来自居群间,8.132%来自居群内,居群间的基因交流较少。
由于贵州居群的psbA-trnH序列与其他居群有较大差异,因此,对其他16个居群以及贵州居群与其周边省区居群的遗传多样性进行了分析,结果显示:其他16个居群的核苷酸多样性指数为0.015 68± 0.000 77,居群间遗传分化系数为0.837 19,基因流为0.10;贵州居群与其周边省区(四川、云南、广西、湖南和湖北)居群的核苷酸多样性指数为0.047 99± 0.010 88,居群间遗传分化系数为0.937 62,基因流为0.03。说明其他16个何首乌野生居群的整体遗传多样性水平偏低;贵州居群与其周边省区居群的遗传分化系数大于其他16个居群,基因交流则小于其他16个居群,说明何首乌野生居群的遗传多样性整体水平在很大程度上受贵州居群的影响。

表3 何首乌17个野生居群间的遗传距离1)Table 3 Genetic distance among seventeen wild populations of Fallopia multiflora(Thunb.)Harald.1)
2.4 何首乌居群的聚类分析
以萹蓄为外类群,依据遗传距离、采用邻接法(NJ)对何首乌17个野生居群进行聚类分析并构建遗传关系树,结果见图1。由图1可看出:17个何首乌野生居群的85个单株可聚成4支。浙江、江西、湖北、福建和四川居群的所有单株,山东居群的4号单株,上海居群的2号和4号单株及云南居群的2~5号单株聚在一起成为第1支,自展支持率达57%,其中,四川居群的5个单株和云南居群的2~5号单株又独立成支,与其他居群分离,但自展支持率仅为54%。广东和湖南居群的所有单株及广西居群的1~4号单株聚在一起成为第2支,自展支持率达86%。第3支包括安徽、甘肃、河南、江苏和陕西居群的所有单株,云南居群的1号单株,广西居群的5号单株,上海居群的1号、3号和5号单株及山东居群的1~3号和5号单株,自展支持率达99%,其中安徽居群相对独立。第4支是与其他居群遗传关系较远的贵州居群的5个单株,自展支持率为99%,与其他居群明显分离,但与外类群关系相对较近。
通过比较可见:何首乌野生居群的聚类结果与psbA-trnH序列分析的划分结果基本一致。
3 讨论和结论
遗传多样性是生物在长期进化和发展过程中形成的自然属性,其不仅体现在种群间和种群内,也体现在不同个体间,是种群和个体遗传变异的总和[15]。特定物种的遗传多样性是该物种长期生存、进化和适应的结果[16]。一般来说,自交物种的核苷酸多样性指数平均值为0.51,异交物种则小于0.1[17],而何首乌野生居群的核苷酸多样性指数仅0.028 56。Buso等[18]认为遗传分化系数大于0.25则表示居群间的分化程度很大,而本研究中何首乌居群间的遗传分化系数达到0.918 68。Slatkin[19-20]将基因流分为高(大于或等于1.000)、中(0.250~0.990)、低(0.000~0.249)3个等级,本研究中何首乌居群间的基因流为0.04,属于低等级。因而,总体上看何首乌野生居群整体遗传多样性水平偏低,居群间遗传分化很大,但居群间的基因流远小于标准,说明遗传漂变是影响何首乌居群遗传结构的主导因素[21]。而何首乌居群间的基因流不足以完全抵制由遗传漂变而引起的居群分化,这也是何首乌居群遗传多样性水平偏低和居群间遗传分化系数较大的原因。

图1 基于psbA-trnH序列分析的何首乌17个野生居群的NJ系统树Fig.1 NJ phylogenetic tree of seventeen wild populations of Fallopia multiflora(Thunb.)Harald.based on psbA-trnH sequence analysis
研究结果表明:除贵州居群外,供试的其他16个居群间的基因流为0.10,遗传分化系数为0.837 19;而贵州居群与其周边省区(四川、云南、广西、湖南和湖北)居群间的基因流仅为0.03,遗传分化系数却达到0.937 62,明显大于其他16个居群间。推测造成这一现象的原因可能是由于云贵高原具有较特殊的喀斯特地貌,由此形成的地理隔离阻碍了基因交流,导致分化较大。因此,何首乌居群的整体多样性水平在很大程度上受贵州居群的影响。从形态上看,17个居群的何首乌外部形态特征基本一致,但贵州居群植株主、侧叶脉的白斑程度最大,叶窦凹陷程度最小,花序长度最短,叶长宽比和宿存花被长度相对较小,开花期相对较晚(另文发表)。贵州居群在形态上的差异得到了psbA-trnH序列分析及聚类分析(自展支持率99%,与供试的其他16个居群明显分离)结果的支持,证明贵州居群确实与其他居群存在较大差异,表现出特殊的形态和遗传特性。推测贵州居群可能是何首乌的1个新变种,但还需多学科研究验证。
聚类分析结果表明:17个何首乌野生居群中有13个居群各自的5个单株聚在一起,仅山东、云南和广西居群各有1个单株与同居群大部分单株较远而被聚到其他分支中,而上海居群的2号和4号单株聚在第1支中,1号、3号和5号单株则聚在第3支中。说明各居群不同单株基于psbA-trnH序列的遗传背景基本一致,仅有少量变异,这一现象可能与这些居群单株间分布的地理环境生态因子不同有关。在进化发展过程中同种不同个体所承受的生境选择存在一定差异,使个体在各自的自然环境下发生了变异。有研究者在黄独(Dioscorea bulbiferaL.)遗传多样性研究中也观察到这一现象[22],因而,同一居群不同个体间存在变异的情况并不少见。
[1]WU Z Y,RAVEN P H.Flora of China:Vol.5[M].Beijing: Science Press,2003:277-350.
[2]国家药典委员会.中华人民共和国药典:2010年版(一部)[M].北京:中国医药科技出版社,2010:164-165.
[3]王凌晖,曹福亮,汪贵斌,等.何首乌野生种质资源的RAPD指纹图谱构建[J].南京林业大学学报:自然科学版,2005,29 (4):37-40.
[4]严寒静,房志坚,余世孝.不同种源何首乌的ITS序列分析及其亲缘关系研究[J].西北植物学报,2008,28(5):922-927.
[5]孙华钦,罗科,邹文俊,等.穿龙薯蓣、黄山药和盾叶薯蓣psbA-trnH片段序列分析[J].应用与环境生物学报,2006,12 (6):792-797.
[6]邵世光,韩丽,马艳红,等.枫斗类石斛cpDNApsbA-trnH的序列分析与鉴别[J].药学学报,2009,44(10):1173-1178.
[7]李永,杨攀,李永华.伏牛山区连翘遗传多样性研究[J].西北植物学报,2011,31(4):665-670.
[8]DOYLE J J,DOYLE J L.A rapid DNA isolation procedure for small quantities of fresh leaf tissue[J].Phytochemical Bulletin,1987,19 (1):11-15.
[9]KUMAR S,TAMURA K,JAKOBSEN I B,et al.MEGA2: molecularevolutionarygeneticsanalysissoftware[J].Bioinformatics,2001,17(12):1244-1245.
[10]KIMURA M.A simple method for estimating evolutionary rates of basesubstitutionsthroughcomparativestudiesofnucleotide sequences[J].Journal of Molecular Evolution,1980,16(2): 111-120.
[12]NEI M.Molecular Evolutionary Genetics[M].New York: Columbia University Press,1987.
[13]LYNCH M,CREASE T J.The analysis of population survey data on DNA sequence variation[J].Molecular Biology and Evolution,1990,7(4):377-394.
[14]FELSENSTEIN J.Confidence limits on phylogenies:an approach using the bootstrap[J].Evolution,1985,39(4):783-791.
[15]中国科学院生物多样性委员会.生物多样性研究的原理与方法[M].北京:中国科学技术出版社,1994.
[16]文亚峰,韩文军,吴顺.植物遗传多样性及其影响因素[J].中南林业科技大学学报,2010,30(12):80-87.
[17]HAMRICK J L,GODT M J W.Allozyme diversity in plant species[M]∥BROWN A H D,CLEGG M T,KAHLER A L,et al.Plant Population Genetics,Breeding and Genetic Resources.Sunderland: Sinauer Associates,Inc.,1989:43-63.
[18]BUSO G S C,RANGEL P H,FERREIRA M E.Analysis of genetic variability in South American wild rice populations(Oryza glumaepatula)with isozymes and RAPD markers[J].Molecular Ecology,1998,7(1):107-117.
[19]SLATKIN M.Estimating levels of gene flow in natural populations[J].Genetics,1981,99(2):323-335.
[20]SLATKIN M.Rare alleles as indicators of gene flow[J].Evolution,1985,39(1):53-65.
[21]WALKER C W,VILC,LANDA A,et al.Genetic variation and population structureinScandinavianwolverine(Gulogulo) populations[J].Molecular Ecology,2001,10(1):53-63.
[22]郑玉红,夏冰,杭悦宇,等.黄独遗传多样性研究[J].西北植物学报,2006,26(10):2011-2017.
(责任编辑:佟金凤)
Genetic diversity of wild populations of Fallopia multiflora based on psbA-trnH analysis
BAI Ming-ming,SUN Xiao-qin,GUO Jian-lin,LI Mi-mi,HANG Yue-yu①(Jiangsu Province Key Laboratory for PlantEx-situConservation,Institute of Botany,Jiangsu Province and the Chinese Academy of Sciences,Nanjing 210014,China),J.Plant Resour.&Environ.2012,21(2):36-44
Sequence ofpsbA-trnH of 85 individuals in 17 wild populations ofFallopia multiflora(Thunb.) Harald.from different provinces and regions in China was amplified and analyzed,and on the basis,genetic diversity among populations was analyzed and cluster analysis of 85 individuals was also carried out by NJ method.The results show that the length ofpsbA-trnH sequence of 85 individuals is 384 bp,in which,there are 167 bp variable sites and 53 bp parsimony informative sites,accounting for 43.5%and 13.8%of the total length of sequence,respectively.Variable types are mainly base deletion and substitution.Variable sites mainly concentrate in the region of 235-281 bp.17 populations are almostly divided into three types according to site variation status.The genetic distances among 17 populations are 0.000-0.172,in which,genetic distances between Guizhou population and other 16 populations are 0.167-0.172,and those among other 16 populations are 0.000-0.017.Nucleotide diversity index (Pi),coefficient of gene differentiation(Nst)and gene flow(Nm)among 17 populations are 0.028 56,0.918 68 and 0.04,respectively.Pi,Nst andNm among other 16 populations except Guizhou population are 0.015 68,0.837 19 and 0.10,respectively.AndPi,Nst andNm between Guizhou population and its neighboring populations(Sichuan,Yunnan,Guangxi,Hu’nan and Hubei)are 0.047 99,0.937 62 and 0.03,respectively.On NJ phylogenetic tree,17 populations are clustered into four branches and individuals tested in most populations are clustered in a same branch and only Guizhoupopulation is clustered alone in a branch,which is basically same with the deviation result by sequence analysis.It is suggested that 91.868%of overall genetic variation of 17 wild populations exists among populations and 8.132%within populations,and gene exchange among populations is less.Except Guizhou population,overall genetic diversity level among other 16 populations is low,indicating that the overall diversity level ofF.multiflorapopulations is affected by Guizhou population at a large extent.
Fallopia multiflora(Thunb.)Harald.;population;psbA-trnH sequence;genetic diversity; NJ phylogenetic tree
book=2012,ebook=77
Q946-33;S567.23+9
A
1674-7895(2012)02-0036-09
2011-12-27
国家农业部DUS测试品种信息DNA测试技术研究项目(200903008-02-05)
白明明(1987—),女,山东郯城人,硕士研究生,主要从事药用植物资源的研究。
①通信作者E-mail:hangyueyu@21cn.com
