超高效液相色谱法测定人参中人参皂苷Re 的含量
2012-08-06孙全乐蔡广知贡济宇长春医学高等专科学校长春130031长春中医药大学长春130117
孙全乐,蔡广知,贡济宇#(1.长春医学高等专科学校,长春130031;.长春中医药大学,长春130117)
美国Waters公司在2004年推出了一种新的液相色谱技术——超高效液相色谱(UPLC)[1],它采用1.7 μm粒径的色谱柱填料,能获得更高的柱效,并且在更宽的线速度范围内使柱效保持恒定,因而有利于提高流动相流速、缩短分析时间、提高分析通量。通过性能优越的色谱柱,精确梯度控制的超高压液相色谱泵,低扩散、低交叉污染的自动进样系统及高速检测器,使UPLC的峰容量、分析效率、灵敏度较常规高效液相色谱(HPLC)[2~4]有了很大的提高,为复杂体系的分离分析提供了良好的平台。
人参皂苷是人参中的主要活性成分,已确定结构的人参皂苷至少在30种以上[5,6]。近年来,人们围绕着人参皂苷的提取、分离、结构鉴定、含量测定、药理等方面展开了深入细致的研究,而人参皂苷的含量测定方法是重点研究课题之一。目前常用的HPLC法虽可达到分离效果,但此法耗时过长、溶剂用量大、成本高。因此,笔者采用UPLC法对人参中人参皂苷Re的含量进行测定,并与HPLC法进行比较,旨在确定更高效、快速和准确的测定方法,为后续研究打下基础。
1 仪器与试药
UPLC仪,含ACQUITY UPLC紫外检测器、Empower工作站(美国Waters公司);KQ-2200E型超声波提取器(昆山市超声仪器有限公司,功率:100 W,频率:50 kHz)。
人参药材购于长春市各药房(吉林大药房、吉深大药房、永新大药房、吉林宏检公司、普天大药房、仁德大药房),经长春中医药大学张雅芝教授鉴定均为真品;人参皂苷Re对照品(中国食品药品检定研究院,批号:110810-200609);乙腈为色谱纯,水为重蒸水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Waters patened hybrid particle细内径柱(50 mm×2.1 mm,1.7 μm);流动相:乙腈-水(19∶81);流速:0.4 mL·min-1;柱温:30 ℃;检测波长:203 nm。
2.2 对照品溶液的制备[7]
精密称取人参皂苷Re对照品适量,加甲醇制成浓度为0.2 mg·mL-1的溶液,摇匀,即得。
2.3 供试品溶液的制备
精密称取人参粗粉5 g,加8倍量水浸泡12 h,放入超声波提取器中,30℃下超声提取20 min,共提取3次,合并提取液,抽滤,水液用水饱和正丁醇萃取5次(20、20、20、15、15 mL),合并正丁醇液,用1%NaOH洗涤2次,每次20 mL,弃去碱液,再用正丁醇饱和水洗涤3次(20、20、15 mL),弃去水液,正丁醇液水浴蒸干,残渣加甲醇溶解并转移至5 mL量瓶中,加甲醇稀释至刻度,摇匀,用0.22 μm微孔滤膜滤过,即得。
2.4 含量测定方法
采用外标法测定。分别吸取对照品溶液和供试品溶液5 μL注入UPLC仪,记录保留时间和峰面积积分值。
2.5 方法学考察
2.5.1 检测波长的选择 取人参皂苷Re对照品溶液在190~240 nm波长范围内作光谱扫描,结果发现人参皂苷Re在203 nm波长处有最大吸收,故选择203 nm作为检测波长。
2.5.2 流动相的选择 考察以不同体积比的乙腈-水(40∶60、30∶70、20∶80、19∶81)体系作为流动相的试验结果,发现当乙腈-水的体积比为19∶81时,人参皂苷色谱峰峰形稳定、无脱尾、分离完全,故最终确定以乙腈-水(19∶81)作为试验用流动相。
2.5.3 专属性考察 取人参皂苷Re对照品溶液、阴性对照溶液(甲醇)和供试品溶液按上述色谱条件分别进样分析,结果显示阴性对照溶液在人参皂苷Re峰保留时间处无干扰峰,人参皂苷Re峰达到有效分离,理论板数按人参皂苷Re峰计算应为6 000,表明本方法专属性良好。
2.5.4 线性关系考察 取人参皂苷Re对照品20.04 mg,置10 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,分别取3.0、4.0、5.0、6.0、7.0 μL,按上述色谱条件进样测定。以峰面积积分值(Y)为纵坐标,质量浓度(X)为横坐标,进行线性回归,得回归方程为Y=9 029X-16 060.2(r=0.999 8,n=5)。结果表明,人参皂苷Re的质量浓度在0.531~1.239µg·mL-1范围内与峰面积积分值呈良好线性关系。
2.5.5 精密度试验 精密吸取人参皂苷Re对照品溶液5 μL,在上述色谱条件下重复进样6次,记录色谱图及峰面积积分值。结果,RSD=1.14%(n=6),表明仪器精密度良好。
2.5.6 稳定性试验 取同一供试品溶液适量,分别于配制后0、0.5、1、2、3、5 h注入UPLC仪测定,记录色谱图及峰面积积分值。结果,RSD=1.03%(n=6),表明室温下供试品溶液在5 h内稳定。
2.5.7 加样回收率试验 精密称取已知含量的样品适量,精密加入一定量的人参皂苷Re对照品(1∶1),按“2.3”项下方法制备供试品溶液,照上述方法测定样品含量,并计算加样回收率,结果见表1。
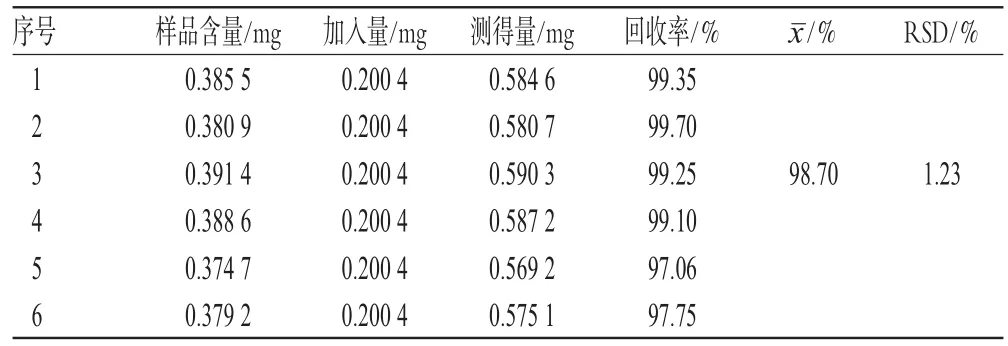
表1 加样回收率试验结果(n=6)Tab 1Results of recovery test(n=6)
2.5.8 重复性试验 取同一批样品适量,共6份,精密称定,分别按“2.3”项下方法制得供试品溶液,精密吸取5 μL注入UPLC仪。结果,人参皂苷Re含量的RSD=0.08%(n=6),表明本方法重复性良好。
2.6 样品含量测定结果
取在不同药店购买的人参药材样品适量,分别按“2.3”项下方法制备供试品溶液,吸取5 μL注入UPLC仪,记录色谱图并计算人参皂苷Re的含量,结果见表2。
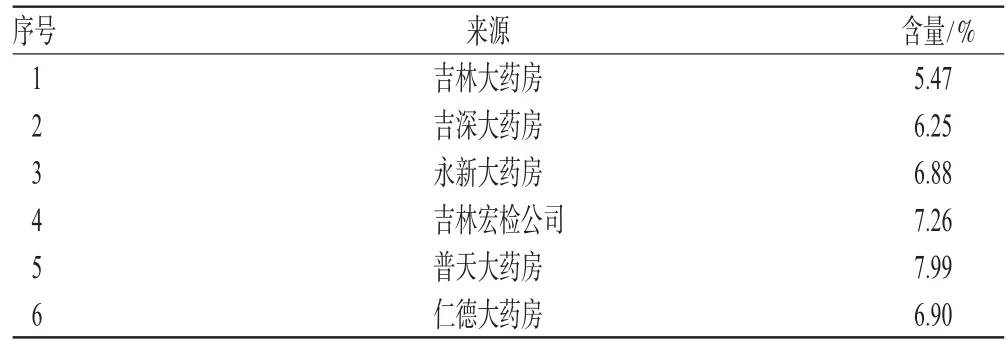
表2 样品含量测定结果(n=3)Tab 2Results of content determination of the sample(n=3)
3 讨论
3.1 UPLC法与HPLC法的比较
与HPLC法比较,UPLC法有几个优点:超高分离度、超高速度、超高灵敏度,具体如下:
3.1.1 缩短分析时间 中药材的成分十分复杂,需要高效和快速的分离分析方法测定其复杂组成。常规HPLC法测定人参皂苷Re的含量一般需要1 h左右的时间,样品组分才能得到较好的分离(见图1);而采用UPLC法只需一半甚至更短的时间就能得到更好的分离效果和分析结果,测定人参皂苷只需在10 min内就可完成试验(见图2),如采用梯度洗脱的方式,还可进一步缩短分析时间[8]。
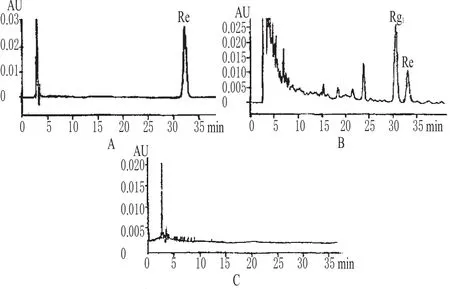
图1 高效液相色谱图(引用文献[8])A.人参皂苷Re对照品;B.供试品;C.阴性对照Fig 1 HPLC chromatograms(referred literatures[8])A.ginsenoside Re control;B.test sample;C.negative control
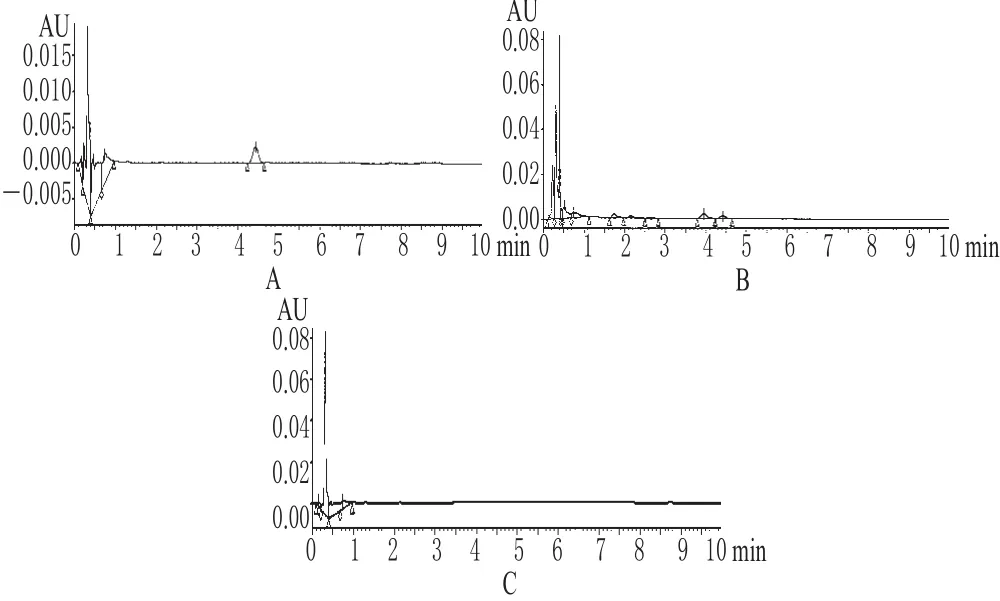
图2 超高效液相色谱图A.人参皂苷Re对照品;B.供试品;C.阴性对照Fig 2 UPLC chromatogramsA.ginsenoside Re control;B.test sample;C.negative control
3.1.2 增加峰容量,提高灵敏度 梯度条件下UPLC的峰容量变化已有报道[8~10],该报道以小分子药物为研究对象,认为流动相比例、梯度洗脱时间、流速、色谱柱长度对峰容量影响最大,短柱、快速梯度变化可获得很高的峰容量;或增加分析时间,即使使用长柱也会获得更高的峰容量。UPLC使用1.7 μm粒径的色谱柱填料,使色谱柱的柱效极大提高、色谱峰变窄、峰容量增加。如图1和图2中,UPLC图中前5 min内共有5个色谱峰达到完全分离,而HPLC图中前40 min内共有6个色谱峰分离完全。此外,UPLC进样量为5 μL时,最高峰的吸收值为0.20 AU;HPLC进样量为5 μL时,与UPLC中最高峰的对应吸收值为1.70 AU,经过换算可得UPLC的灵敏度是HPLC的7倍左右。UPLC的高灵敏度优势对微量组分的研究具有重要意义。
3.2 方法转换
在保证相同分离度的条件下,UPLC与HPLC可以进行简单的方法转换,使HPLC上50 min完成的分析在UPLC上不到8 min即可完成。在相同分离机制条件下,主要转换流速、进样量及流动相梯度条件:根据转换软件,HPLC上10 μL的进样量在UPLC上为5 μL,HPLC上1 mL·min-1的流速转换到UPLC上为0.21 mL·min-1,从而使在HPLC上50 min完成的分析在UPLC上用21.78 min即可完成;在仪器压力允许及保证分离度的前提下,UPLC可适当增大流速,最大可达0.6 mL·min-1,在此条件下,7.62 min即可完成分析。本试验应用此转换方法,原HPLC转换为UPLC的条件为进样量5 μL、流速0.21 mL·min-1,可在10 min内完成分析;当增大流速为0.4 mL·min-1时,测试时间可缩短至5 min。
3.3 小结
本试验采用UPLC法测定人参中人参皂苷Re的含量,供试品溶液在10 min内分离完全,检测时间比HPLC法缩短了30 min,可见UPLC能够更快、更好地完成以往HPLC的工作。UPLC不但可以节省时间、提高效率、减少溶剂的消耗,还能为质谱提供最佳的色谱条件,为中药的分析建立良好的平台,为信息库的建立奠定良好的基础。有理由相信,UPLC法的超高分析速度、超高灵敏度,必将会在复杂中药体系的分离测试领域开创崭新局面。
[1]金高娃,章飞芳,薛兴亚.超高效液相色谱在复杂体系中药分离分析中的应用[J].世界科学技术中医药现代化,2006,8(3):110.
[2]刘 军,王燕恒,付承光.高效液相色谱法分析人参皂苷[J].药物分析杂志,1998,18(2):132.
[3]Zhai WM,Yuan YS,Zhou YX,et al.HPLC fingerp-rints identification ofPanax ginsengC.A.Mey.P.quinque folinL.andP.notoginseng(Burk.F.H.Chen)[J].Chin J Chin Mater Mater Med,2001,26(7):481.
[4]高 敏.人参注射剂质量标准研究[J].云南中医中药杂志,2003,24(5):31.
[5]张均田,刘 云,屈志炜,等.人参皂苷Rb1和Rg1对小鼠中枢神经递质受体和脑内蛋白质合成的影响[J].药学学报,1988,23(1):12.
[6]王本祥.人参研究进展[M].天津:天津科学技术出版社,1991:22-54.
[7]国家药典委员会.中华人民共和国药典(一部)[S].2010年版.北京:中国医药科技出版社,2010:8.
[8]张雪光,俄丽丹,孙 巍,等.高效液相色谱法测定人参片中人参皂苷Rg1和人参皂苷Re的含量[J].中国药业,2003,12(10):42.
[9]刘 飞,贺浪冲.人参质量控制的定性与定量方法研究[J].药物分析杂志,2002,22(3):173.
[10]徐莲英,陶建生,冯 怡,等.中药制剂发展的回顾[J].中成药,2000,22(1):6.
