高钙铬渣及其解毒后可溶六价铬含量分析方法研究
2012-04-04王天贵李强秦利玲吕书玲
王天贵,李强,秦利玲,吕书玲
(1.河南工业大学化学化工学院,河南郑州450001;2.山西大同大学化学与化工学院)
综述与专论
高钙铬渣及其解毒后可溶六价铬含量分析方法研究
王天贵1,李强2,秦利玲1,吕书玲1
(1.河南工业大学化学化工学院,河南郑州450001;2.山西大同大学化学与化工学院)
分析了铬渣中六价铬含量的测定方法、六价铬的浸取方法、解毒铬渣的评价方法等几个方面的问题。通过对比国内外大量的研究结果,指出了铬渣中六价铬浸取评价方法存在的问题。由于铬渣的强碱性及其强大的酸中和能力,所谓的酸液浸取大多名不符实,用硫酸、硝酸、盐酸稀溶液浸取铬渣中六价铬要比USEPA Method 3060A碱消解方法效果差很多;用HJ/T 299—2007和HJ/T 300—2007评价解毒后的铬渣存在很多问题;用GB 5085.3—2007评价铬渣的浸出毒性也有不严密的地方。测定铬渣中六价铬最好用物理方法,例如XANES方法;浸取铬渣中的六价铬宜采用USEPA Method 3060A的碱消解方法。
铬渣;六价铬;浸取;解毒评价
铬盐是常用的基础化工原料,其生产起始于铬铁矿的高温碱融氧化,传统的高钙生产工艺产生大量废渣,简称铬渣(或高钙铬渣)。铬渣中含有高达2%(质量分数)左右的六价铬,而六价铬是国际公认的致癌物,因此铬渣就成了危害极大的危险固体废物。由于铬渣物相及化学成分复杂,使得其解毒处理极其困难、代价很高。虽然国内外早已认识到铬渣的危害,并持续不断地进行铬渣处理研究[1-4],至今仍没有好的办法。尽管西方发达国家早已淘汰了高钙焙烧工艺,但是其历史遗留的高钙铬渣仍具有巨大的潜在威胁,因此英美环保机构及相关学者仍在致力于铬渣解毒研究[5-14]。中国是铬盐生产大国,历史上遗弃了大量高钙铬渣。虽然国家早就明令淘汰高钙焙烧工艺,但至今无法完全杜绝铬渣的产生。多年来,国内相关政府部门、企业、科研机构、大专院校等单位进行了大量的科研和实践,提出了许多铬渣解毒处理及综合利用的办法,也获得了不少的成果,但至今铬渣的威胁依旧。要彻底解决铬渣污染问题还有很长的路要走。
1 铬渣的主要物相成分
铬渣的化学成分主要是铁、铝、镁、钙、硅的氧化物,属强碱性(pH大于12)。铬渣的物相组成很复杂,在露天堆存或雨水冲刷后还可能发生变化,但主要物相组成不外乎那么十来种[1-4,14],参见表1。多数研究者认为,铬渣中的六价铬主要嵌入在钙矾石、水铝钙石、水榴石几种晶体中,但是不同研究者得出的六价铬在几种矿相中的比例有差异。当然也有人认为六价铬存在于水滑石[Mg6Al2(CO3)(OH)16·4H2O]中,而不是水铝钙石中[3,6]。考虑到铬渣成分的复杂性以及铬渣来源、年代的很大差异,分析结果有一些差异也很正常。由于六价铬镶嵌在矿物的晶格中,还包裹在高温焙烧形成的结核中,使得六价铬的溶出或还原都极其困难,这是铬渣难以彻底解毒的关键所在。目前,解毒铬渣常用两种方法,一种是将六价铬原位还原成三价铬固定下来,另一种是将六价铬浸出,回收利用或进一步还原成三价铬。由于铬渣是强碱性,在碱性环境中六价铬氧化能力很弱,再加上还原剂很难渗透接触六价铬,因此第一种方法难以彻底解毒铬渣。第二种方法面临同样的问题,并且要将铬渣全部酸溶将会消耗大量的酸,而碱溶无法将六价铬全部溶出。
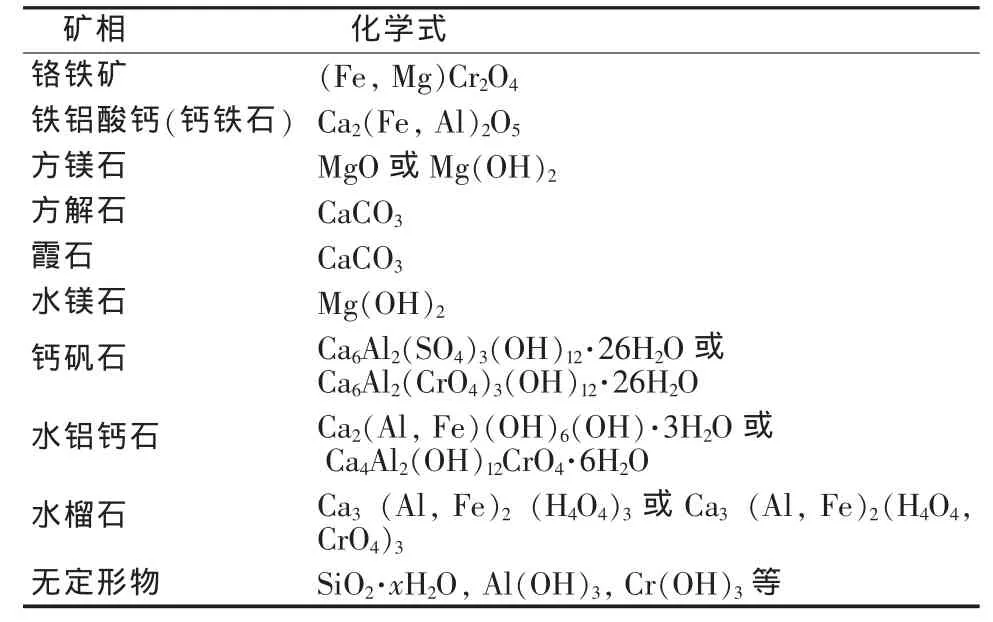
表1 铬渣的主要物相成分[1-4,14]
2 确定铬渣中的总六价铬
对于刚产生的新鲜铬渣,确定其六价铬含量并不难。可以假定其不含还原性物质,采用强酸将其完全溶解,然后测定溶液中六价铬浓度,计算铬渣中六价铬含量。当前,新鲜铬渣已经不多,更常见的是陈旧性铬渣。对于陈旧铬渣,尤其是多年露天堆放、填埋,与雨水、地下水、污水长期接触的铬渣,由于混入还原性物质,不能采用酸溶的方法浸出铬。因为酸性条件下六价铬氧化能力很强,很容易被还原。最好的办法是采用物理的方法确定铬渣中的六价铬,比如国外常用的X射线近边能谱方法(XANES)[1,15-18]。该方法不仅可以避免六价铬还原,可以同时确定六价铬和三价铬含量,也不破坏样品的物相结构。XANES不仅可以分析各种固体矿物六价铬含量,还可以分析土壤、各种生物质中六价铬含量。这种方法在国外已使用多年,而国内至今未见有人使用。
3 铬渣中的六价铬浸取
物理方法确定铬渣中六价铬尽管有很多优点,但至今应用并不普及。一是仪器昂贵,二是分析结果要借助专用软件进行处理,多数实验室并不具备这样的条件。最常用的方法还是溶液浸取,但既要保证六价铬浸取彻底,又要保证结果不失真,确实有难度。国内有不少文章谈到铬渣的六价铬浸取研究,总的结果是:六价铬浸出量与浸取液pH、液固比、固体粒度、固液接触时间和方式、浸取温度、辅助浸取手段等因素有关,其中最主要的因素是浸取液的pH。事实上,由于铬渣属强碱性,主要由碱性氧化物组成,因此铬渣具有很强的中和酸能力。根据国外的研究结果,每千克干铬渣能够中和8~13 mol的氢离子[5]。很多所谓的酸浸取研究,事实上都是在碱性条件下完成的。以HJ/T 299—2007方法为例,以液固比为10 mL/g、pH为3.2的硫酸硝酸混合水溶液浸取铬渣中的六价铬,只要液固一接触,浸取液马上变成强碱性。因此用该方法浸取铬渣中的六价铬完全违背该方法的设计初衷,跟用水浸取没有什么差别。英国、美国主要关心历史遗留铬渣对土壤和地下水的污染问题,研究对象多为含有还原性物质的陈旧性铬渣,因此除了采用物理方法(如XANES)外,确定铬渣或铬渣污染的土壤中的六价铬普遍采用美国环保部的碱溶方法(USEPA Method 3060A)。大量研究表明,除非用浓酸将铬渣的物相破坏,使得绝大部分氧化物溶解,可保证六价铬全部溶出,否则,稀酸水溶液即使与铬渣长时间接触也只能溶出部分六价铬。J.G.Famer等[1]和J.M.Tinjum等[5]都证明,用稀酸水溶液与铬渣接触,只有加酸量能够维持浸取液pH在8左右时,六价铬的溶出量最大,酸量的进一步增加或减少都会使得溶出的六价铬减少。J.G.Famer等采用盐酸作为六价铬浸取剂,接触时间长达26 d,结果表明,最多只能浸取总六价铬的20%左右。J.M.Tinjum等采用硝酸和硫酸作为六价铬浸取剂,接触时间为72 h,浸取液中六价铬浓度与文献[1]的盐酸浸取结果近似,但最多只有USEPA Method 3060A浸取量的30%左右。此外,文献[5]还简单说明了六价铬浓度随pH变化的原因。即:在pH为8~12时,溶液中六价铬浓度随pH降低而增加是由于含铬矿相的溶解以及吸附态六价铬的脱附;在pH为5~8时,溶液中六价铬浓度随pH降低而减小是由于铬渣颗粒表面转化为正电荷,以阴离子形态存在的六价铬被吸附,原先溶解的部分矿物二次沉淀,包裹夹带部分六价铬,导致溶液中六价铬浓度降低;pH小于5以后,矿物会随酸浓度增加继续溶解,导致溶液中六价铬浓度增加。
由于铬渣的强碱性及其巨大的中和酸能力,顺序浸取土壤中六价铬的各种方法[7]不适用于铬渣,因为用于浸取不同结合形态六价铬的浸取液都会成为强碱性而失去其原有意义。况且,土壤中的那些所谓的形态在铬渣中根本就不存在。
4 铬渣解毒效果的评价方法
“铬渣污染治理环境保护技术规范(暂行)”(HJ/ T 301—2007)是一部专门针对高钙铬渣管理的行业标准,其中详细规定了有关铬渣存放、解毒处理、综合利用等各个方面的要求。在该标准的“铬渣的解毒”部分并没有明确规定解毒后铬渣中六价铬含量的指标要求,而是在“铬渣的综合利用”和“铬渣的最终处置”部分规定了六价铬的浸出要求。除了规定进入生活垃圾填埋场的铬渣使用HJ/T 300—2007方法浸取评价六价铬含量外,其余的铬渣都要求使用HJ/T 299—2007方法浸取评价六价铬含量。HJ/T 299—2007和HJ/T 300—2007方法的本意都是希望用稀酸溶液浸取六价铬,而将其应用于解毒后的铬渣却会产生问题。下面分别针对两种不同的解毒铬渣进行说明。
4.1 干法解毒铬渣
铬渣的干法解毒是指用还原剂在高温下将铬渣中的六价铬还原成三价铬固定下来。通常情况下,干法解毒除了还原部分六价铬外,并不改变铬渣的其他属性,尤其是不会改变其强碱性及高pH缓冲能力。即使是像HJ/T 301—2007说的,铬渣经高温还原后经硫酸亚铁水溶液水淬降温处理,其属性也不会有明显变化。J.G.Farmer等[10]实验过用70倍空隙体积(相当于当地2 a的降雨量)的NaCl稀水溶液冲洗5 cm厚的铬渣柱层,溶液的pH仅仅从11.8下降到11.5。前已述及,HJ/T 299—2007使用的是pH为3.2的硫酸硝酸混合水溶液作浸取剂,这种浸取剂接触到铬渣马上会被中和,实际上相当于用水进行浸取。HJ/T 300—2007使用pH为2.64的醋酸水溶液作浸取剂,可想而知,结果和HJ/T 299—2007不会有多大差别。表2为对比实验结果(铬渣来自河南某铬盐厂)。
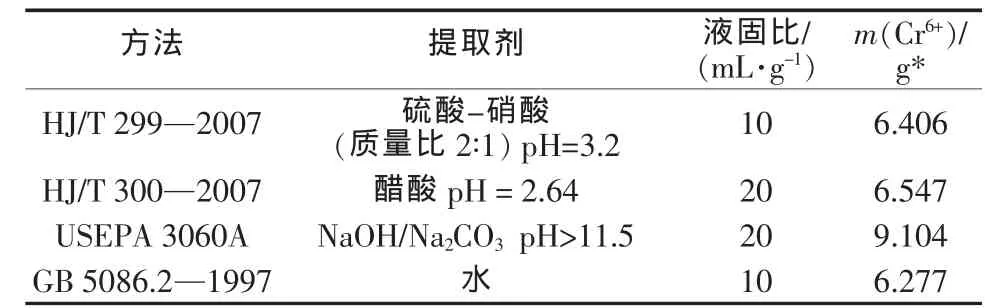
表2 不同浸取方法六价铬量
4.2 湿法解毒铬渣
除了干法解毒,另一种常用的铬渣解毒方法是将铬渣加入水(或酸碱水溶液)中,然后添加还原剂(铁屑、亚铁、多硫化钙、各种生物质等)将铬渣中的六价铬还原为三价铬,经此方法处理的铬渣统称为湿法解毒铬渣。由于湿法解毒多在酸性条件下进行,因此湿法解毒铬渣一般不再具有强碱性和强缓冲能力,但会残留夹带还原剂。不再具有强碱性和强缓冲能力并不意味着没有碱性和缓冲能力,这还要看具体的解毒处理条件。因此pH为3.2的硫酸硝酸混合水溶液和pH为2.64的醋酸水溶液仍有可能被迅速中和。假如解毒后的铬渣真的失去了碱性和缓冲能力,六价铬浸取在pH为3.2和pH为2.64下进行,则会造成结果失真。众所周知,在酸性环境中六价铬有很强的氧化能力,将10倍的酸液与铬渣充分接触18 h,必将造成铬渣中残余六价铬的进一步浸出和还原,导致结果偏低。对于解毒过程中pH偏高(比如5~7)、还原速率低且还原剂难与铬渣分离(比如各种生物质残渣、细菌)的情况,这种问题会更加突出。在有还原剂存在下,不仅浸取过程中会发生六价铬还原,在分光光度法测定六价铬浓度时,加酸酸化更会加剧六价铬还原。这种含有还原剂的湿法解毒铬渣,除了前面提到的物理方法外,目前似乎还没有其他的方法能够准确地评价其六价铬含量。如果一定要浸取,使用碱性溶液比酸性溶液更合理,毕竟六价铬在碱性环境中氧化能力要弱很多,结果失真的概率也更低。
综上所述,HJ/T 299—2007和HJ/T 300—2007方法类似于USEPA Method 1311,不适宜用于铬渣、解毒铬渣及受铬渣污染的土壤中六价铬的浸取评价,建议使用USEPA Method 3060A的碱性消解法,或者GB 5086.1—1997的水浸取法。
“危险废物鉴别标准 浸出毒性鉴别”(GB5085.3—2007),“适用于任何生产、生活和其他活动中产生固体废物的浸出毒性鉴别”。在该标准的“鉴别标准”部分,也规定用HJ/T 299—2007浸取固体废物,但在其“实验方法”中提到“六价铬及其化合物的样品的前处理方法参照附录T”,附录T也就是USEPA Method 3060A。如果铬渣先按 USEPA Method 3060A碱消解,然后再按HJ/T 299—2007浸取其六价铬,则不仅可以使六价铬得到充分浸取,也可以最大程度上避免其在浸取过程中被还原,方法设计比较科学。但美中不足的是,该标准只是说“六价铬及其化合物的样品的前处理方法参照附录T”,但并没有说含有六价铬的固体废物都必须进行这样的前处理,更没有说碱消解后的消解液怎么处理(是单独计量、单独分析、单独计算还是和HJ/T 299—2007的浸取液一起分析计算)、消解后的固体要不要洗涤、要不要计量?是直接加HJ/T 299—2007的抽提剂还是洗涤、干燥、称量后再按液固比要求添加抽提剂?这几个方面都是需要再明确的。
[1] Farmer J G,Paterson E,Geelhoed J S,et al.Identification and geochemical modeling of processes controlling leaching of Cr(VI)and other major elements from chromite ore processing residue[J].Geochimica et Cosmochimica Acta,2002,66(22):3927-3942.
[2] Hillier S,Roe M J,Geelhoed J S,et al.Role of quantitative mineralogical analysis in the investigation of sites contaminated by chromite ore processing residue[J].The Science of the Total Environment,2003,308(1/2/3):195-210.
[3] Chrysochoou M,Fakra S C,Marcus M A,et al.Microstructural analyses of Cr(VI)speciation in chromite ore processing residue(COPR)[J].Environ.Sci.Technol.,2009,43(14):5461-5466.
[4] Hillier S,Lumsdon D G,Brydson R,et al.Hydrogarnet:a host phase for Cr(VI)in chromite ore processing residue(COPR)and other highpHwastes[J].Environ.Sci.Technol.,2007,41(6):1921-1927.
[5] Tinjum J M,Benson C H,Edil T B.Mobilization of Cr(VI)from chromite ore processing residue through acid treatment[J].Science of The Total Environment,2008,391(1):13-25.
[6] Chrysochoou M,Dermatas D.Application of the Rietveld method to assess chromium(Ⅵ)speciation in chromite ore processing residue[J].Journal of Hazardous Materials,2007,141(2):370-377.
[7] Elzinga E J,Cirmo A.Application of sequential extractions and X-ray absorption spectroscopy to determine the speciation of chromium in Northern New Jersey marsh soils developed in chromite ore processing residue(COPR)[J].Journal of Hazardous Materials,2010,183(1/2/3):145-154.
[8] Dermatas D,Chrysochoou M,Moon D H,et al.Ettringite-induced heave in chromite ore processing residue(COPR)upon ferrous sulfatetreatment[J].Environ.Sci.Technol.,2006,40(18):5786-5792.
[9] Moon D H,Wazne M,Dermatas D,et al.Long-term treatment issues with chromite ore processing residue(COPR):Cr6+reduction and heave[J].JournalofHazardousMaterials,2007,143(3):629–635.
[10] Farmer J G,Paterson E,Bewley R J,et al.The implications of integrated assessment and modelling studies for the future remediation of chromite ore processing residue disposal sites[J].Science of the Total Environment,2006,360(1/2/3):90-97.
[11] Cao J,Zhang W X.Stabilization of chromium ore processing residue(COPR)with nanoscale iron particles[J].Journal of Hazardous Materials,2006,132(2/3):213-219.
[12] Graham M C,Farmer J G,Anderson P,et al.Calcium polysulfide remediation of hexavalent chromium contamination from chromite ore processing residue[J].Science of the Total Environment,2006,364(1/2/3):32-44.
[13] Moon D H,Wazne M,Jagupill S C,et al.Particle size and pH effects on remediation of chromite ore processing residue using calcium polysulfide(CaS5)[J].Science of The Total Environment,2008,399(1/2/3):2-10.
[14] 纪柱.铬渣长期堆存后的组成变化及对治理的影响[J].铬盐工业,2007(2):1-11.
[15] Szulczewski M,Helmke P,Bleam W.Comparison of XANES analyses and extractions to determine chromium speciation in contaminated soils[J].Environ.Sci.Technol.,1997,31(10):2954-2959.
[16] Strub E,Plarre R,Radtke M.Determination of Cr(Ⅵ)in wood specimen:A XANES study at the Cr K edge[J].Nuclear Instruments and Methods in Physics Research B,2008,266(10):2405-2407.
[17] Wei Yuling,Lee Y C,Hsieh H F.XANES study of Cr sorbed by a kitchen waste compost from water[J].Chemosphere,2005,61(7):1051-1060.
[18] Peterson M L,Brown G E,Parks G A,et al.Differential redox and sorption of Cr(Ⅲ/Ⅵ)on natural silicate and oxide minerals:EXAFS and XANES results[J].Geochimica et Cosmochimica Acta,1997,61(16):3399-3412.
联系方式:tgwang2006@126.com
Evaluation of methods for determination of hexavalent chromium contained in chromite ore processing residue
Wang Tiangui1,Li Qiang2,Qin Liling1,Lü Shuling1
(1.School of Chemistry and Chemical Engineering,Henan University of Technology,Zhengzhou,450001,China 2.School of Chemistry and Chemical Engineering,Shanxi Datong University)
Hexavalent chromium (Cr6+)determination,and extraction protocols from chromite ore processing residue(COPR),and the evaluation of the harmlessness measures of COPR were reviewed.Problems of some Cr6+leaching protocols of COPR were pointed out by referring to quite a lot of related publications at home and abroad.As the strong alkali and the great acid neutralization capability of COPR,many so-called Cr6+acid solution extractions are untrue.USEPA Method 3060A leaches much more Cr6+than the diluted aqueous solutions of sulfuric acid,nitric acid or hydrochloric acid.HJ/T 299—2007 and HJ/T 300—2007 methods can not determine genuine Cr6+content of the treated COPR.GB 5085.3—2007 has flaws for evaluating the leaching toxicity of COPR.Best choice for determining Cr6+content of COPR is physical analysis,such as X-ray absorption near-edge structure analysis;and it′s better to leach Cr6+from COPR by USEPA Method 3060A.
chromite ore processing residue;hexavalent chromium;leaching method;detoxification evaluation
TQ136.11
:A
:1006-4990(2012)04-0001-04
2011-10-26
王天贵(1962—),男,博士,教授,主要从事清洁生产研究,已发表论文60多篇,曾获河南省自然科学优秀论文奖。
