解决氧化还原酶反应体系中辅酶问题的策略及其应用
2012-02-26江金鹏吴旭日陈依军
江金鹏,吴旭日,陈依军
中国药科大学生命科学与技术学院 化学生物学研究室,江苏 南京 210009
氧化还原酶是一类重要的生物催化剂。与化学催化剂相比,其催化的氧化还原反应具有反应条件温和、立体选择性强、副反应少和产物得率高等优点,约占所有生物催化反应的30%以上,并且随着新酶的不断发现和酶理性改造技术的发展,这一比例还在逐年升高[1-2]。虽然氧化还原酶能选择性还原含羰基和醛类化合物为手性氨和手性醇[3-4],并且这些产物可作为重要手性模块在手性药物制备、精细化工品生产及手性材料合成等多个领域具有巨大的应用前景,但是至今真正应用于工业生产的案例却屈指可数。其根本的制约因素在于:1) 氧化还原酶需要辅酶的参与,而这些辅酶大多为烟酰胺类,主要是NAD+/NADH和NADP+/NADPH;2) 在工业生物催化应用中,通常需要人为添加外源性辅酶以完成催化反应,而烟酰胺类辅酶价格昂贵、稳定性差、难以重复利用,导致反应成本高昂。因此,如何降低辅酶用量和成本成为氧化还原酶工业化应用的一个重要课题。
经过较长时间的研究,人们已建立了多种方法用于提高辅酶的利用效率,降低反应中辅酶的成本,这些方法主要包括:采用酶法、电化学法、光化学法或化学法原位再生辅酶;利用代谢工程等手段改造细胞的代谢途径,提高内源性辅酶的含量和利用率;研究辅酶替代物等。这些策略都能够不同程度地改善反应体系中氧化还原酶的活性和辅酶的稳定性,实现辅酶及其替代物的重复利用,提高氧化还原反应的效率,降低生产成本,从而推动了氧化还原酶反应体系的工业化进程。
1 辅酶的体外循环再生
1.1 酶法循环再生
辅酶的酶法循环再生是指利用酶促反应实现还原态或氧化态辅酶的重复利用。由于操作简单可控、选择性高、稳定性好、再生体系与反应体系兼容性强以及共底物价格低廉等优点,目前仍是研究较多和使用较为广泛的方法。根据实施策略的不同,酶法循环再生系统又分为两类,第一类是底物偶联法 (图1),即在反应体系中仅有一种氧化还原酶,该酶既能催化共底物再生辅酶,又能利用辅酶和底物合成产物。目前最为常用的底物偶联系统有异丙醇 (IPA)-NADPH循环系统和乙醇-NADH循环系统。例如,美国Codexis公司以IPA作为共底物构建了IPA-NADPH辅酶再生体系,为氧化还原酶KRED催化合成抗哮喘药物孟鲁司特的手性醇中间体连续提供质子供体,使底物完全转化成产物的最大浓度达到100 g/L[5]。虽然底物偶联法简化了反应体系并提高了辅酶的利用效率,但是该方法具有明显的缺点:1) 仅利用一种酶在同一体系中催化两个不同方向的反应,因此较难获得最适反应的热力学条件;2) 过量的共底物也会对酶产生抑制作用而降低催化效率。
另一类为酶偶联法 (图1),即在一个反应体系中联合应用两种氧化还原酶催化不同反应,一个酶催化底物反应获得产物,另一个酶催化共底物反应实现辅酶再生。该方法由于具有共底物价廉、反应的热力学条件容易控制、辅酶再生效率高和循环稳定性好等优点,因此已成为应用最为广泛的辅酶再生技术。目前常用的酶偶联辅酶循环系统有:甲酸-甲酸脱氢酶 (FDH) 系统、葡萄糖-葡萄糖脱氢酶 (GDH) 系统、6-磷酸葡萄糖-6-磷酸葡萄糖脱氢酶系统以及亚磷酸-亚磷酸脱氢酶 (PTDH) 系统。例如,本实验室从不动杆菌Acinetobacter baylyi ATCC 33305克隆了一种新的氧化还原酶,命名为双羰基还原酶(DKR)[6-7],构建了DKR和FDH的偶联体系用于合成他汀类药物手性二醇中间体。通过进一步引入水-正己烷两相系统,该偶联体系的底物浓度提高至105 g/L[8],较大地降低了生产成本。
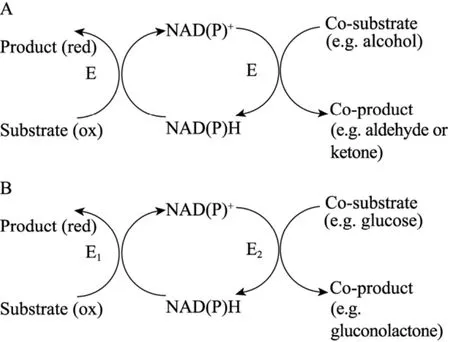
图1 辅酶的酶法循环再生Fig.1 Cofactor regeneration by enzymatic reactions. (A) Substrate coupling method. E: enzyme for catalyzing two different reactions in one system. (B) Enzyme coupling method. E1: product-preparing enzyme; E2: cofactor regeneration enzyme.
除了在反应体系中添加氧化还原酶及相应的底物进行辅酶的循环再生外,利用微生物细胞内天然存在的氧化还原酶也能实现辅酶的循环再生。Yang等[9]发现南极酵母 Rhodotorula sp. AS2.224细胞粗提液中含有内源性甘露醇脱氢酶,并利用该酶再生NADPH,进而辅助羰基还原酶催化底物2-乙酰基吡啶的还原,转化率高达96.8%,ee值大于99%。最近,Yan等[10]构建了羰基还原酶SCR1的大肠杆菌工程菌,证实了该菌的粗酶液中存在多种可用于NADPH再生的内源性氧化还原酶。经反应条件优化后,含有羰基还原酶SCR1的粗酶液能催化80 mmol/L的α-羟基苯乙酮的还原,转化率高达90.1%,ee值大于99%,NADPH的总转化数为3 604。此外,本实验室也考察了大肠杆菌细胞内源性葡萄糖脱氢酶和葡萄糖-6-磷酸脱氢酶再生辅酶的效率,发现补加葡萄糖能有效利用内源性氧化还原酶和DKR形成的偶联体系催化合成他汀类药物手性二醇中间体,其催化效率与DKR-FDH偶联反应体系相当[11]。这些实验结果表明,利用微生物细胞自身的氧化还原酶来实现辅酶再生的方法不仅能够降低生产成本,而且使实验操作进一步简化,是一种有效的辅酶再生策略。但是,由于大多数天然存在的氧化还原酶存在表达量低、催化活性弱以及容易失活等缺点,其工业化应用的潜力并不大。
1.2 电化学法、光化学法和化学法循环再生
电化学法是利用电极提供或接受电子使还原态或氧化态辅酶再生的方法。该方法虽然具有反应体系简单、不需外加第二个酶和共底物、成本低以及无副产物等优点,但也存在辅酶再生速率低、酶易失活、需要提供高电压和电极易钝化等诸多问题。尽管电极表面改性和新电子介体的寻找能够在一定程度上解决上述问题,例如Kim等[12]制备的具有氧化还原性质的氧化锡电极能够有效再生辅酶,将其与醇脱氢酶偶联可以用于合成丙酮,但电极材料本身的性能局限导致该方法目前尚处于实验室研究阶段,需要继续发展和完善。
光化学法采用光敏染料 (如亚甲蓝) 经可见光照射激发后作为电子载体提供电子给氧化态辅酶而实现辅酶的再生。虽然该方法具有绿色环保、价格低廉等优点,但过于复杂的反应体系和低下的辅酶再生效率致使其至今还停留在实验室研究阶段,离实际应用还有一段距离。
辅酶的化学再生法就是利用化学电子供体如连二亚硫酸盐将电子传递给氧化态的辅酶而实现NAD(P)H的再生。其中,利用廉价的氢气以及性能优良的钌复合物分别作为最终还原剂和催化剂还原氧化态辅酶是一种富有潜力的辅酶再生系统[13]。然而,该系统的辅酶再生效率偏低与稳定性差等缺点不利于工业化应用,有待进一步研究。
2 细胞代谢与内源性辅酶
2.1 内源性辅酶再生系统的构建
在不影响微生物细胞自身正常代谢的情况下,利用基因工程技术向细胞内引入外源性氧化还原酶如GDH、PTDH和FDH,构建胞内辅酶再生系统以实现辅酶的循环利用是解决多酶、多底物辅酶再生系统兼容性和复杂性的有效手段。Berríos-Rivera等[14]将FDH引入大肠杆菌,加入甲酸后细胞内NADH的含量增加了1倍,乙醇脱氢酶催化合成乙醇的效率也随之提高了15倍。针对体外辅酶再生体系效率低、操作繁琐与稳定性差等缺点,本实验室构建了DKR和GDH的共表达重组大肠杆菌,通过酶催化反应动力学分析和对工程菌细胞内辅酶浓度的实时监测,发现在全细胞催化反应过程中,当其他影响反应效率的因素均相同时细胞内辅酶浓度决定了全细胞的催化效率。在此规律指导下,通过优化反应条件,在不需外加辅酶的情况下,利用以上共表达菌株催化底物完全转化为合成他汀手性侧链的中间体——3R,5S-双羟基酯化合物,且反应体系中底物浓度比单纯表达dkr基因的细胞提高了15倍[15],有效地解决了辅酶供应这一制约因素,为氧化还原酶的工业化应用提供了新的思路。然而,该方法并不能提高细胞内辅酶的总量,有时甚至需要添加外源性辅酶来增加细胞内辅酶的浓度以加速催化反应的完成。
2.2 培养基成分的优化
由于培养基组分对微生物次生代谢途径影响较大,因此优化培养基的成分,尤其是碳源,在一定程度上能直接调节氧化态和还原态辅酶的含量及其比例。San等[16]向培养基中添加多种氧化形态的碳源 (葡萄糖,山梨醇和葡萄糖酸),发现不同碳源导致大肠杆菌内 NADH/NAD+的比例产生较大差别,其原因在于不同氧化形态的碳源参与的糖酵解过程存在差异。此外,Liu等[17]在发酵培养基中加入 NAD+的生物合成前体烟酸 (NA) (图2),发现8 mg/L NA能够有效促进细胞内NAD+的合成,从而增加了光滑球拟酵母Torulopsis glabrata细胞内产物丙酮酸的浓度。
2.3 辅酶合成途径的改造
微生物细胞内的辅酶生成和降解始终处于一个动态的平衡过程,NAD (P) 的合成途径主要分为从头合成途径 (De novo pathway) 和补救合成途径 (Salvage pathway) (图2),其中改造补救途径是增加内源性辅酶总量的常用手段。烟酸磷酸核糖转移酶 (NAPRTase) 催化 NA磷酸化生成烟酸单核苷酸 (NAMN) 的反应是补救途径的限速步骤。据此,Witholts等[18]对NAPRTase的结构和功能进行了系统研究,并将其在大肠杆菌中过量表达,使内源性烟酰胺辅酶的总量提高了5倍。Berrios-Rivera等[19]通过测定过量表达NAPRTase工程菌内的烟酰胺辅酶的含量,同样证实了高活力的 NAPRTase能够促进内源性辅酶的合成,并且NADH/NAD+比例随NAPRTase活性的增加而降低。
在辅酶的补救途径中,NAD+激酶 (NADK)催化NAD+发生磷酸化即可生成NADP+[20-21]。Lee等[22]在胸苷生产菌中过量表达了NADK后,内源性NADPH的含量增加到原先的1.3倍,胸苷的产率也随之提高1.2倍。最近,另一新发现的NADK在构巢曲霉Aspergillus nidulans中被过量表达同样能够使内源性NADPH总量得到明显提高[23]。
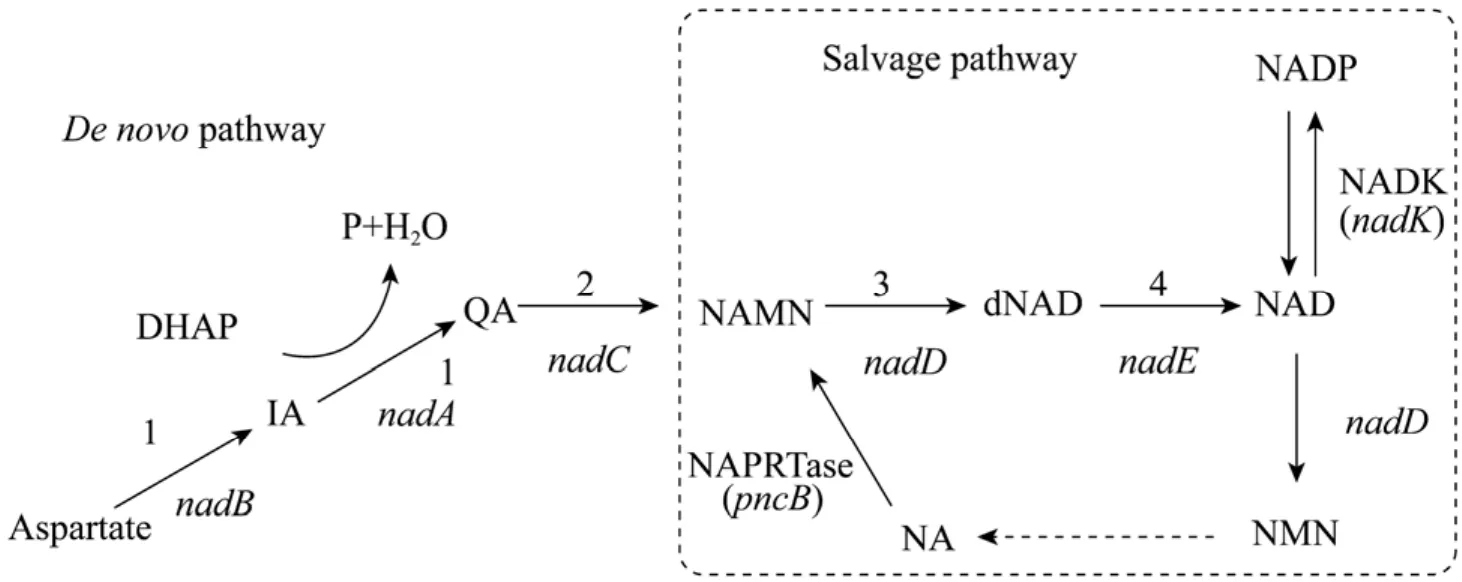
图2 辅酶的从头合成途径与补救途径Fig. 2 Cofactor NAD (P) biosynthesis through the de novo pathway and salvage pathway. 1: quinolinic acid (QA) sythetase; 2: QA phosphoribosyltransferase; 3: nicotinic acid monocleotide (NAMN) adenylyltransferase; 4: NAD sythetase. NADK: NAD+ kinase; NAPRTase: nicotinic acid phosphoribosyltransferase; DHAP: dihydroxyacetone phosphate; IA: iminoaspartate; dNAD: desamido NAD; NA: nicotinic acid; NMN: nicotinamide mononucleotide.
2.4 辅酶再生代谢途径的调控
细胞内的代谢网络由众多复杂的代谢途径所构成,通过对代谢网络中辅酶再生和代谢途径的调控可以改变内源性辅酶的存在形态和利用度。例如磷酸戊糖途径 (PPP) 是再生还原态辅酶NADPH的主要代谢途径,Poulsen等[24]通过在黑曲霉Aspergillus niger内过量表达PPP中的6-磷酸葡萄糖脱氢酶,使内源性NADPH的浓度增加了2至9倍。葡萄糖-6-磷酸脱氢酶 (G6PDH)是PPP途径中另一种生成NADPH的关键酶,通过在大肠杆菌中过量表达 G6PDH,可以使内源性NADPH/NADP+的比例提高6倍[25]。
虽然通过对 PPP的改造可以有效增加内源性NADPH的含量,但提高NADH的细胞内浓度则需要修饰其他代谢途径,如甲醇氧化途径。Pscheidt等[26]系统分析了毕氏酵母 Pichia pastoris的甲醇氧化途径中 3个关键酶 (乙醇氧化酶、甲醛脱氢酶 (FLD) 和FDH) 的动力学性质,通过模拟和实验发现 FLD是该代谢途径中NADH再生的主要限制酶。因此,在P. pastoris中过量表达FLD就可达到有效再生NADH的目的。此外,在厌氧发酵过程中,乳酸脱氢酶催化丙酮酸还原产生乳酸,同时消耗大量的NADH。为避免这一过程的发生,通过构建过量表达甘油脱氢酶和丙酮醛合酶的乳酸脱氢酶缺陷型突变株,不仅增加了内源性 NADH含量,而且利用该菌株催化合成 1,2-丙二醇的产率也提高了67%[27]。
虽然人们通过运用各种策略使细胞的代谢流分布发生改变以达到提高内源性辅酶总量并调节NAD (P)+/NAD (P) H比例的目的,但是由于细胞内的代谢网络非常复杂,至今仍缺乏对辅酶相关代谢途径的全面认识,精确调控内源性辅酶的含量及其形态还比较困难。因此,很有必要对胞内辅酶的代谢情况进行更深入的研究。最近,通过在大肠杆菌内过量表达NAD (H) 跨膜转运蛋白NTT4并敲除NAD+合成基因nadE,获得了能直接利用细胞外NAD (H) 的NAD+营养缺陷型工程菌株,利用该菌株测定了大肠杆菌内辅酶的浓度阈值[28]。这一研究结果进一步加深了人们对细胞内辅酶代谢情况的认识,也将有助于对内源性辅酶的含量和NAD (P)+/NAD (P) H比例进行更精确的调控,为有效设计辅酶依赖型氧化还原酶介导的全细胞反应体系提供了新的手段。
3 辅酶替代物的研究和应用
针对天然辅酶稳定性差、价格昂贵以及回收利用效率低等缺点,合成具有类似功能的替代物可能是解决辅酶现存问题的一条新途径。
经过长期的努力,人们已经合成了众多辅酶结构类似物,希望从中筛选到理想的天然辅酶替代物。迄今,研究较多的是一类含有烟酰胺结构的三嗪类染料,即活性蓝2 (C.I. Reactive Blue 2,图3)。由于该类染料能与氧化还原酶活性中心的辅酶结合位点相互作用,因此Burton等通过对众多三嗪类染料的衍生物进行筛选,获得了具有天然辅酶活性的化合物活性蓝 N-3 (Blue N-3)[29](图3)。但是,活性蓝N-3结构中的蒽醌生色团在水溶液中的溶解度较差,并且因其在紫外-可见光范围内具有较大吸收而不利于用光谱法监测反应过程。为了解决这些问题,活性蓝N-3中的蒽醌生色团被替换成萘等基团,由此合成了一系列活性蓝 N-3的衍生物[30-31],并从中筛选到结构简单、水溶性高、稳定性好以及紫外-可见光吸收值低的化合物CL4[32](图3),这使辅酶替代物研究又进了一步。然而,与天然辅酶相比,这类替代物的催化活性仍然较低。因此,如何进一步优化结构,提高催化活性,减少反应体系中的用量是该类辅酶替代物研究的重点。
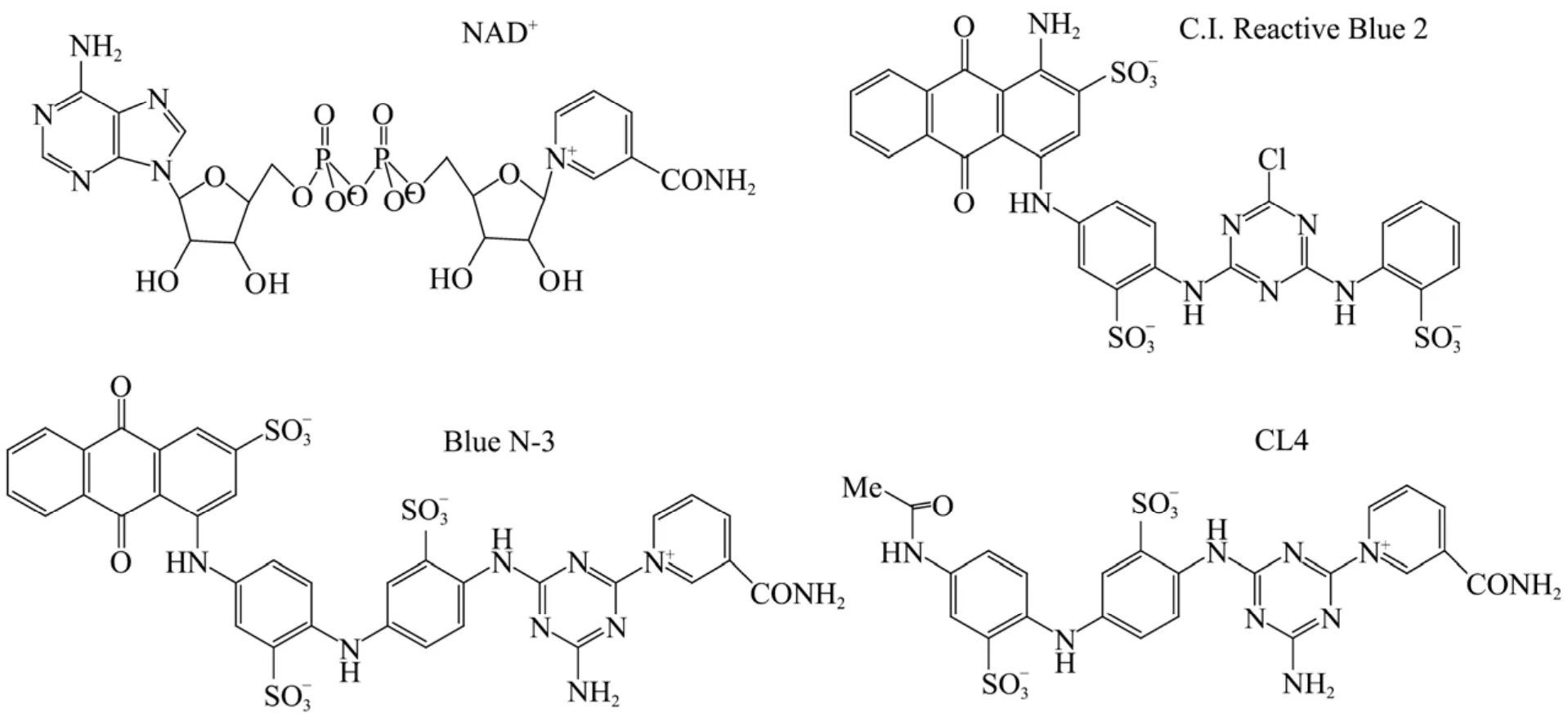
图3 NAD+,三嗪类染料活性蓝2及辅酶替代物活性蓝N-3,CL4的结构式Fig. 3 Chemical structures of NAD+, triazine dyes C.I. Reactive Blue 2, Blue N-3 and CL4.
辅酶依赖型细胞色素P450具有重要的催化特性,因此针对该类酶催化的氧化还原反应人们设计了多种辅酶替代体系[33-35],其中采用含钴的化合物 (Cobalt sepulchrate) (图4) 作为辅酶替代物为细胞色素P450提供电子的体系最具代表性和最为典型[36]。Faulkner等[37]利用电化学法与cobalt sepulchrate偶联为细胞色素P450连续提供电子,从而使P450催化月桂酸发生ω-羟化作用,并且反应速率与NADPH介导的P450体系相当。在此基础上,为了改善水溶性和提高利用效率,Udit等[38]合成了亲水性的NADPH替代物1,1’-二羧基二茂钴 (图4)。此外,为了进一步简化反应体系和降低成本,Schwaneberg等用锌粉作为电子供体构建了zinc:cobalt (III) sepulchrate系统。将P450 BM-3与这一新系统偶联,其催化合成对硝基苯酚的效率达到了NADPH反应体系的20%[39]。虽然众多研究表明,cobalt sepulchrate是一类较有潜力的辅酶替代物,但是反应过程中产生的过氧化物以及体系中存在的氧气会影响cobalt sepulchrate和P450的活性,因此需要进一步研究对该体系进行优化。
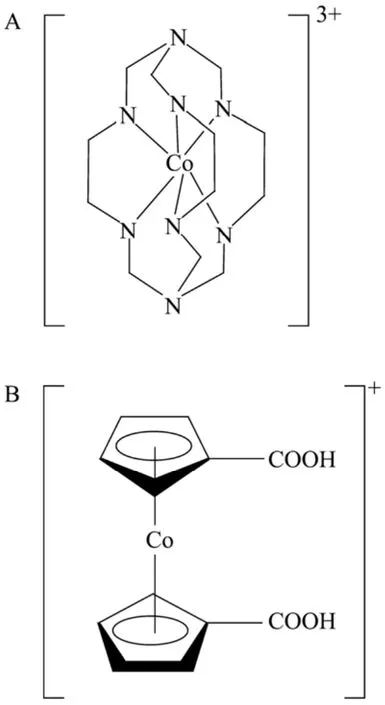
图4 钴复合物的结构式Fig. 4 Structures of cobalt complexes. (A) Cobalt sepulchrate. (B) 1,1’-dicarboxycobaltocene cation.
1-苯甲基-1,4-二氢烟酰胺是另一类重要的NAD+结构类似物。将其与[Cp*Rh (bpy) (H2O)] (OTf)2偶联,可以构建辅酶替代物化学再生系统。Lo等[40]利用该系统辅助马肝醇脱氢酶催化合成手性芳香和烷基取代醇,产率达到 90%,ee值大于90%。随后,该系统被应用于2-羟基-3-单加氧酶 (HbpA) 合成邻苯二酚[41]以及P450突变体 (W1064S,R966D) 催化对硝基苯氧基癸酸的羟化反应[42]。由此可见,1-苯甲基-1,4-二氢烟酰胺作为某些氧化还原酶的辅酶替代物进行相应的催化反应具有良好的应用前景。但是,该替代物仍然存在活性低和水溶性差等问题。因此,今后的工作还需集中在寻找新的辅酶结构类似物,并对现有的辅酶替代物及与之匹配的氧化还原酶进行结构改造,以改善现有替代物的各种缺陷。
4 总结与展望
虽然氧化还原酶展现出巨大的应用前景并在化学工业绿色化的进程中发挥着重要作用,但至今真正用于工业化生产的案例却屈指可数。其根本的制约因素是:绝大多数氧化还原酶需在烟酰胺类辅酶的参与下才能完成催化反应,而烟酰胺类辅酶价格昂贵、稳定性差并且难以重复利用,造成催化反应成本高昂。尽管近几十年来人们针对辅酶问题已提出了多种解决方法,如构建辅酶循环再生系统、改造辅酶合成途径、调控辅酶再生代谢途径以及设计辅酶替代物等,并且取得了一些实质性进展,但是这些方法大多都存在各种各样的问题和缺陷,严重阻碍了氧化还原酶在实际工业化生产中的应用。
基于辅酶工程的研究现状和地球能源的逐渐枯竭以及人类环保意识的日益增强,采用理性设计或随机筛选的方法获得能够直接利用水分子或氢气作为电子供体完成氧化还原反应的独特氧化还原酶,以及研究和完善有效的辅酶供应体系,将有可能从根本上解决辅酶问题,由此破解限制氧化还原酶工业化应用的瓶颈,进一步扩大氧化还原酶的应用范围,真正实现重要化学品的绿色化工业制造。
[1] Straathof AJJ, Panke S, Schmid A. The production of fine chemicals by biotransformations. Curr Opin Biotechnol, 2002, 13(6): 548−556.
[2] DeSantis G, Davis BG. The expanding roles of biocatalysis and biotransformation. Curr Opin Chem Biol, 2006, 10(2): 139−140.
[3] Huang Y, Liu N, Wu XR, et al. Dehydrogenases/ Reductases for the synthesis of chiral pharmaceutical intermediates. Curr Org Chem, 2010, 14(14): 1447−1460.
[4] Chen YJ, Chen C, Wu XR. Dicarbonyl reduction by single enzyme for the preparation of chiral diols. Chem Soc Rev, 2012, 41(5): 1742−1753.
[5] Liang J, Lalonde J, Borup B, et al. Development of a biocatalytic process as an alternative to the (−)-DIP-Cl-mediated asymmetric reduction of a key intermediate of montelukast. Org Process Res Dev, 2010, 14(1): 193−198.
[6] Wu XR, Liu N, He YM, et al. A bacterial enzyme catalyzing double reduction of a β, δ-diketo ester with unprecedented stereoselectivity [EB/OL]. [2011-10-20]. Nature Precedings, 2008, http://hdl. handle.net/10101/npre. 2008.1697.1.
[7] Wu XR, Liu N, He YM, et al. Cloning, expression, and characterization of a novel diketoreductase from Acinetobacter baylyi. Acta Biochim Biophys Sin, 2009, 41(2): 163−170.
[8] Wu XR, Chen C, Liu N, et al. Preparation of ethyl 3R, 5S-6-(benzyloxy)-3, 5-dihydroxy-hexanoate by recombinant diketoreductase in a biphasic system. Bioresour Technol, 2011, 102(3): 3649−3652.
[9] Yang W, Xu JH, Pan J, et al. Efficient reduction of aromatic ketones with NADPH regeneration by using crude enzyme from Rhodotorula cells and mannitol as cosubstrate. Biochem Eng J, 2008, 42(1): 1−5.
[10] Yan Z, Nie Y, Xu Y, et al. Biocatalytic reduction of prochiral aromatic ketones to optically pure alcohols by a coupled enzyme system for cofactor regeneration. Tetrahedron Lett, 2011, 52(9): 999−1002.
[11] Wu XR, Wang LL, Wang SZ, et al. Stereoselective introduction of two chiral centers by a single diketoreductase: an efficient biocatalytic route for the synthesis of statin side chains. Amino Acids, 2010, 39(1): 305−308
[12] Kim YH, Yoo YJ. Regeneration of the nicotinamide cofactor using a mediator-free electrochemical method with a tin oxide electrode. Enzyme Microb Technol, 2009, 44(3): 129−134.
[13] Wagenknecht PS, Penney JM, Hembre RT. Transition-metal-catalyzed regeneration of nicotinamide coenzymes with hydrogen. Organometallics, 2003, 22(6): 1180−1182.
[14] Berríos-Rivera SJ, Bennett GN, San KY. Metabolic engineering of Escherichia coli: increase of NADH availability by overexpressing an NAD+-dependent formate dehydrogenase. Metab Eng, 2002, 4(3): 217−229.
[15] Wu XR, Jiang JP, Chen YJ. Correlation between intracellular cofactor concentrations and biocatalytic efficiency: coexpression of diketoreductase and glucose dehydrogenase for the preparation of chiral diol for statin drugs. ACS Catal, 2011, 1(12): 1661−1664.
[16] San KY, Bennett GN, Berríos-Rivera SJ, et al. Metabolic engineering through cofactor manipulation and its effects on metabolic flux redistribution in Escherichia coli. Metab Eng, 2002, 4(2): 182−192.
[17] Liu LM, Li Y, Shi ZP, et al. Enhancement of pyruvate productivity in Torulopsis glabrata: increase of NAD+availability. J Biotechnol, 2006, 126(2): 173−185.
[18] Wubbolts MG, Terpstra P, van Beilen JB, et al. Variation of cofactor levels in Escherichia coli. Sequence analysis and expression of the pncB gene encoding nicotinic acid phosphoribosyltransferase. J Biol Chem, 1990, 265(29): 17665−17672.
[19] Berríos-Rivera SJ, San KY, Bennett GN. The effect of NAPRTase overexpression on the total levels of NAD, the NADH/NAD+ratio, and the distribution of metabolites in Escherichia coli. Metab Eng, 2002, 4(3): 238−247.
[20] Raffaelli N, Finaurini L, Mazzola F, et al. Characterization of Mycobacterium tuberculosis NAD kinase: functional analysis of the full-length enzyme by site-directed mutagenesis. Biochemistry, 2004, 43(23): 7610-7617.
[21] Mori S, Kawai S, Shi F, et al. Molecular conversion of NAD kinase to NADH kinase through single amino acid residue substitution. J Biol Chem, 2005, 280(25): 24104−24112.
[22] Lee HC, Kim JS, Jang W, et al. Thymidine production by overexpressing NAD+kinase in an Escherichia coli recombinant strain. Biotechnol Lett, 2009, 31(12): 1929−1936.
[23] Panagiotou G, Grotkjaer T, Hofmann G, et al. Overexpression of a novel endogenous NADH kinase in Aspergillus nidulans enhances growth. Metab Eng, 2009, 11(1): 31−39.
[24] Poulsen BR, Nøhr J, Douthwaite S, et al. Increased NADPH concentration obtained by metabolic engineering of the pentose phosphate pathway in Aspergillus niger. FEBS J, 2005, 272(6): 1313−1325.
[25] Lim SJ, Jung YM, Shin HD, et al. Amplification of the NADPH-related genes zwf and gnd for the oddball biosynthesis of PHB in an E. coli transformant harboring a cloned phbCAB operon. J Biosci Bioeng, 2002, 93(6): 543−549.
[26] Schroer K, Luef KP, Hartner FS, et al. Engineering the Pichia pastoris methanol oxidation pathway for improved NADH regeneration during whole-cell biotransformation. Metab Eng, 2010, 12(1): 8−17. [27] Berríos-Rivera SJ, San KY, Bennett GN. The effect of carbon sources and lactate dehydrogenase deletion on 1, 2-propanediol production in Escherichia coli. J Ind Microbiol Biotechnol, 2003, 30(1): 34−40.
[28] Zhou YJ, Wang L, Yang F, et al. Determining the extremes of the cellular NAD(H) level by using an Escherichia coli NAD+-auxotrophic mutant. Appl Environ Microbiol, 2011, 77(17): 6133−6140.
[29] Burton SJ, Stead CV, Ansell RJ, et al. An artificial redox coenzyme based on a triazine dye template. Enzyme Microb Technol, 1996, 18(8): 570−580.
[30] Dilmaghanian S, Stead CV, Ansell RJ, et al. Synthesis and properties of a naphthalenecontaining artificial redox coenzyme. Enzyme Microb Technol, 1997, 20(3): 165−173.
[31] Ansell RJ, Dilmaghanian S, Stead CV, et al. Synthesis and properties of new coenzyme mimics based on the artificial coenzyme Blue N-3. Enzyme Microb Technol, 1997, 21(5): 327−334.
[32] Ansell RJ, Small DAP, Lowe CR. Characterisation of the artificial coenzyme CL4. J Mol Catal B: Enzym, 1997, 3(5): 239−252.
[33] Fang X, Halpert JR. Dithionite-supported hydroxylation of palmitic acid by cytochrome P450BM-3. Drug Metab Dispos, 1996, 24(11): 1282−1285.
[34] Vilker VL, Reipa V, Mayhew M, et al. Challenges in capturing oxygenase activity in vitro. J Am Oil Chem Soc, 1999, 76(11): 1283−1289.
[35] Munge B, Estavillo C, Schenkman JB, et al. Optimization of electrochemical and peroxidedriven oxidation of styrene with ultrathin polyion films containing cytochrome P450cam and myoglobin. ChemBioChem, 2003, 4(1): 82−89.
[36] Estabrook RW, Shet MS, Fisher CW, et al. The interaction of NADPH-P450 reductase with P450: an electrochemical study of the role of the flavin mononucleotide-binding domain. Arch Biochem Biophys, 1996, 333(1): 308−315.
[37] Faulkner KM, Shet MS, Fisher CW, et al. Electrocatalytically driven omega-hydroxylation of fatty acids using cytochrome P450 4A1. Proc Natl Acad Sci USA, 1995, 92(17): 7705−7709.
[38] Udit AK, Arnold FH, Gray HB. Cobaltocenemediated catalytic monooxygenation using holo and heme domain cytochrome P450 BM3. J Inorg Biochem, 2004, 98(9): 1547−1550.
[39] Schwaneberg U, Appel D, Schmitt J, et al. P450 in biotechnology: zinc driven ω-hydroxylation of p-nitrophenoxydodecanoic acid using P450 BM-3 F87A as a catalyst. J Biotechnol, 2000, 84(3): 249−257.
[40] Lo HC, Fish RH. Biomimetic NAD+models for tandem cofactor regeneration, horse liver alcohol dehydrogenase recognition of 1, 4-NADH derivatives, and chiral synthesis. Angew Chem Int Ed, 2002, 41(3): 478−481.
[41] Lutz J, Hollmann F, Ho TV, et al. Bioorganometallic chemistry: biocatalytic oxidation reactions with biomimetic NAD+/NADH co-factors and [Cp*Rh(bpy)H]+for selective organic synthesis. J Organomet Chem, 2004, 689(25): 4783−4790.
[42] Ryan JD, Fish RH, Clark DS. Engineering cytochrome P450 enzymes for improved activity towards biomimetic 1, 4-NADH cofactors. ChemBioChem, 2008, 9(16): 2579−2582.
