宏基因组学挖掘新型生物催化剂的研究进展
2012-02-26王魁汪思迪黄睿刘玉焕
王魁,汪思迪,黄睿,刘玉焕
中山大学生命科学学院,广东 广州 510275
宏基因组学起源于上世纪70年代土壤微生物基因组DNA的研究;1980年,Torsvik[1]建立了土壤细菌总DNA的直接提取方法;1991年,Pace研究组[2]通过构建环境样品的DNA文库筛选得到16S rRNA基因 (rDNA),首次证明了从环境中克隆基因的可行性;1995年,Healy等[3]以木质纤维素底物富集培养后的微生物为样品,通过构建基因组文库,筛选得到纤维素酶,证明了从环境样品中直接获得生物催化剂的可行性;1996年,Stein等[4]通过构建海水样品的基因组文库筛选到了一个新的古菌 16S rRNA基因,确立了环境基因组学在挖掘新基因研究中的特殊地位。1998年,Handelsman 等[5]在前人研究的基础上,首次提出了“宏基因组(Metagenome)”的概念并使用了“Metagenomics”这一名词,由此宣告了宏基因组学(Metagenomics) 的诞生。宏基因组是指某一特定生境中全部微生物遗传物质的总和,包含了可培养的和尚不能培养微生物遗传信息的总和。所谓宏基因组学就是利用非培养的分子生物学技术、方法和手段,对宏基因组进行系统研究,即分析微生物在环境中的基因组集合,研究其群落结构、进化关系、相互协作关系与生态功能等。它绕过了微生物分离培养过程,成为研究环境样品中高达 99%的不可培养微生物 (Uncultured microorganism) 的有力手段[6],为后续的筛选提供更加全面和多样的基因资源,有效地提高了新型生物催化剂的筛选效率。宏基因组学从诞生至今仅有十余年时间,但已引起了世界的广泛关注。通过 SCI检索发现,自 2002年以来关于宏基因组的研究报道已经超过1 200篇,而且呈现逐年升温的趋势 (图1)。宏基因组学在新型生物催化剂的研究中取得了令人瞩目的进展,成为国际生命科学技术研究的热点和重点之一。其基本策略流程为:采集样品;提取特定环境中的基因组 DNA;构建宏基因组文库;筛选阳性克隆子;目的基因的亚克隆和表达;目的催化剂的生化特性分析 (图2)。近年来研究者们已利用宏基因组文库技术从不同环境样品中筛选到了脂肪酶/酯酶、淀粉酶、木聚糖酶、纤维素酶、β-葡萄糖苷酶等多种具有工业应用潜力的生物催化剂 (表1)。随着宏基因组学在挖掘新型生物催化剂中的广泛使用,我们进入了宏基因组学挖掘生物催化剂的时代[7]。本文着重从宏基因组学研究的样品来源、宏基因组DNA的提取、宏基因组文库的构建和目标克隆的筛选策略4个关键方面着手,系统综述了采用宏基因组学方法进行新型生物催化剂研究的现状,并对今后的研究和发展方向提出了展望。

图1 近年来宏基因组学研究的报道情况Fig. 1 Research papers on Metagenomics in recent years.
1 宏基因组文库的样品来源
微生物数量巨大,种类繁多,广泛分布于不同的物理化学环境,在自然界长期缓慢的进化过程中,已经产生了功能多样的、在不同生理环境下具有高度精确性和特异性的生物催化剂,适应了各自的环境。所以,自然界的微生物中隐藏了一个巨大的宝藏,蕴藏着大量适于不同工业生产条件 (温度、pH、压力、离子强度和化合物浓度)的生物催化剂,且不同微环境中生物催化剂的种类和生化性质大不相同,而人们一直在寻找的某种生物催化剂很有可能已经存在于某一特定环境的微生物中了[7]。因此,在采用宏基因组学方法挖掘生物催化剂时,样品来源格外重要。随着构建的宏基因组文库类型的日渐增多,Ivanova等[35]认为有必要将宏基因组文库进行分类,以便对不同环境中的基因组信息进行比较分析。迄今为止,为了筛选生物催化剂,研究者们构建了各种各样的环境样品文库,它们的样品来源可归为5大类:陆地环境,水生环境,寄生环境,人工环境及其他特殊环境 (表2)。

图2 宏基因组学策略挖掘生物催化剂的流程图Fig. 2 General process of metagenomic strategies in mining biocatalysts
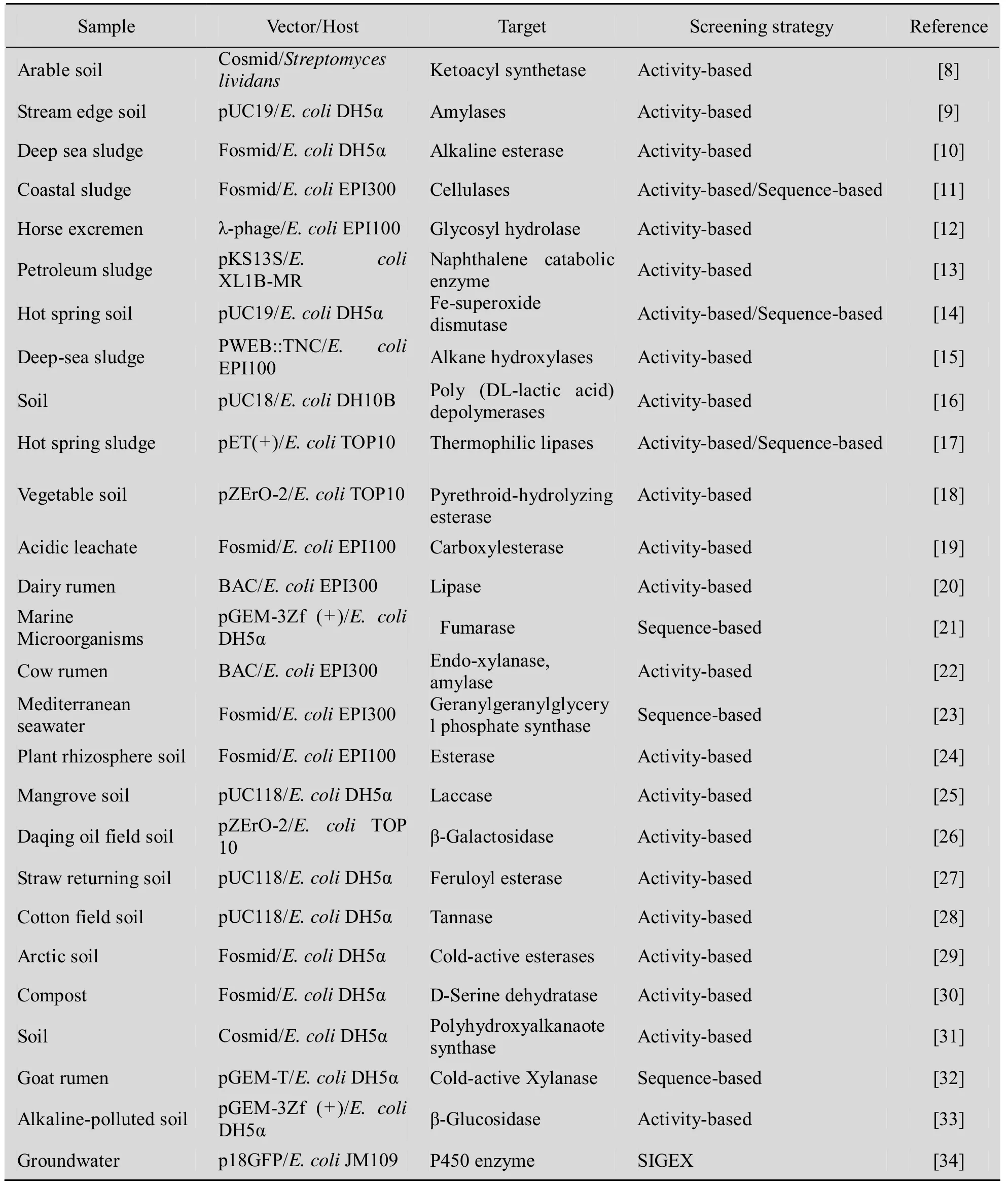
表1 近年来构建的宏基因组文库及筛选到的生物催化剂Table 1 Examples of isolated biocatalysts from metagenomic libraries in recent years
目前,宏基因组学研究的样品主要来源于土壤环境和海洋环境。这是因为土壤具有微生物进行生长繁殖和生命活动所需的各种条件,是微生物生活的最适宜环境。据估计,每克土壤中含有高达10 000种不同的微生物[36],这表明土壤微生物宏基因组是一个巨大的基因资源库,对它们的挖掘能够获得大量的生物酶制剂[37];而海洋环境在压力、盐分、温度和营养成分上的极端变化赋予了海洋微生物在物种资源、基因功能和生态功能上无与伦比的生物多样性。本实验室从红树林、农药污染的棉田、油田等环境的土壤样品中筛选到了酯酶[18]、漆酶[25]、β-半乳糖苷酶[26]、阿魏酸酯酶[27]、单宁酶[28],并在此基础上研究了这些新型生物催化剂的酶学特性。
为了获得性能更加多样的生物催化剂,研究者们需要广泛地采集环境样品,特别是一些极端环境中的样品。

表2 宏基因组文库样品来源的分类Table 2 Classification of samples used for metagenomic library construction
2 宏基因组DNA的提取
宏基因组DNA的提取是构建宏基因组文库的关键步骤,因为环境样品总DNA的浓度、纯度、片段大小和偏好性等因素将直接影响宏基因组文库的质量和代表性。DNA提取方法大体可分为两类:直接提取法和间接提取法。
直接提取法,又称原位裂解法,这类方法是通过物理法、化学法或酶解法直接裂解环境样品中微生物的细胞壁来提取DNA并加以纯化。目前常用的裂解方法中,物理法有反复冻融法、超声法、玻璃珠击打法、液氮碾磨法等;化学法所采用的化学试剂有表面活性剂 (如SDS)、盐类、有机溶剂等;酶解法则主要采用溶菌酶和蛋白酶K。不同裂解法的差别在于细胞破壁的方式不同。直接提取法无需对环境样品中的微生物进行复苏处理,操作简便,且样品中的微生物能够被裂解得比较完全,故DNA提取得率高,所获得的基因组DNA能较好地反映样品中微生物种群的多样性。但由于操作过程中机械剪切力大,导致所获得的 DNA片段较小 (1~50 kb),且多为平端;此外,由于提取的基因组DNA中混有金属离子、腐殖酸、多糖、多酚化合物等杂质,纯度较低,往往还需要经过纯化处理才能满足后续分子生物学操作 (如DNA酶切、PCR扩增) 的需要。该法提取的DNA主要适用于以质粒或λ噬菌体为载体的小片段文库的构建。
间接提取法,又称异位裂解法,是利用梯度离心或者差速离心等方法先把微生物从环境样品中分离出来,然后再用比较温和的方法提取基因组 DNA。该方法受土壤中杂质的污染较小,提取条件比较温和,所获得的DNA纯度较高,片段大 (20~500 kb),适合于构建以柯斯粘粒(Cosmid) 和细菌人工染色体 (Bacterial artificial chromosome,BAC) 为载体的大片段文库;但该方法操作繁琐,前分离过程会造成部分微生物细胞的丢失,加上温和条件下细胞壁较厚的微生物不易被裂解,因此所获得的DNA中基因组信息的广泛性不及原位裂解法,得率只有原位裂解法的1%~10%[37]。
从DNA产率、纯度、片段长度及对整个微生物群落的覆盖率等方面进行评价,直接法和间接法各有优势和不足[38]。总体来看,国内外学者和研究人员采用直接提取法的较多。针对直接提取法得到的环境总DNA中杂质较多的问题,研究者们建立了多种 DNA提取纯化方法。Zhou等[39]通过在基因提取过程中使用PVPP和CTAB去除多糖和腐植酸,获得了适于建库的高质量环境总 DNA,此方法在后来的宏基因组学研究中被广泛采用;Braid等[40]通过絮凝作用有效地去除杂质;Desai等[41]采用温和的裂解方法裂解土壤微生物,然后通过活性炭吸附和离子交换层析两步法有效地去除了土壤中的离子和腐植酸等抑制剂,得到了可用于 PCR反应的高纯度和高浓度土壤总DNA;Pang等[42]针对堆肥环境中高浓度腐殖酸的存在 (堆肥的腐殖酸含量比土壤高10~100倍),使用Sephadex G200+酸洗PVPP层析柱与电洗脱两步纯化的方法纯化堆肥环境来源的DNA,成功地构建了一个含有10万个克隆的柯斯质粒文库,并从这个文库中筛选到一个新的β-葡萄糖苷酶基因。本实验室在Zhou法[39]基础上进行改进,选用商品化的凝胶回收试剂盒对DNA粗提液进行纯化回收,实验操作简单,回收效率高,采用此法构建了棉田土壤宏基因组文库,并从中筛选得到一个具有菊酯农药降解酶活性、阿魏酸酯酶活性和单宁酶活性的酶蛋白的基因[28]。
随着宏基因组学的发展,许多新的DNA提取技术不断被提出,使得人们可以从各种各样的环境样品中获得高质量的总 DNA。然而,并没有一种标准的或通用的环境DNA提取方法。任何DNA提取方法都存在一定的偏好性,导致选择性地富集某些微生物的 DNA,同时选择性地遗漏某些微生物的 DNA,从而影响所提取的DNA的种群覆盖率。在实际应用中,应根据所研究生境的特性和研究目的进行选择和优化。
3 宏基因组文库的构建
3.1 载体的选择
载体在宏基因组技术中具有重要地位,常用载体有质粒 (Plasmid)、细菌人工染色体 (BAC)、柯斯粘粒 (Cosmid)、福斯黏粒 (Fosmid)等,此外还有噬菌体P1克隆系统 (P1-clone)、由P1衍生 的 人 工 染 色 体 (P1-derived artificial chromosome,PAC)、酵母人工染色体 (Yeast artificial chromosomes,YAC)和哺乳动物人工染色体 (Mammalian artificial chromosome,MAC)等 (表3)。目前,在新型催化剂的研究中,所采用的载体有质粒、Cosmid、Fosmid、BAC、λ噬菌体以及各种穿梭载体等。载体的选择应考虑以下因素:研究目的、所提取的环境总DNA的质量、欲插入目的片段的最大长度、所需要的载体拷贝数、欲采用的宿主以及筛选策略。如对腐殖质含量较高或剪切较严重的DNA样品适宜构建质粒文库,而对于含较大基因簇或大片段的DNA样品则适宜构建Cosmid、Fosmid或BAC文库。但是,无论以何种载体构建文库,都必须使文库最大程度地覆盖样本中所有微生物的基因组。
3.2 宿主的选择
宿主的选择应考虑以下因素:所选择的载体类型,宿主的转化效率、重组载体在宿主细胞中的稳定性、宿主能否为相关功能基因提供必需的转录表达体系、对异源表达基因产物是否有较强的相容性、以及目标性状 (如抗菌性) 缺陷型等。目前,构建用于筛选新酶的宏基因组文库时,最常用的宿主为大肠杆菌E. coli,其优点在于操作简单、繁殖迅速、培养代谢易于控制、遗传信息明确。其次是芽胞杆菌 Bacillus、变铅青链霉菌Streptomyces lividans 和 恶 臭 假 单 胞 菌Pseudomonas putida[8,43]。

表3 宏基因组文库常用载体比较Table 3 Comparison of different vectors used in metagenomic library construction
4 目标克隆的筛选
由于环境样品中微生物种类众多,文库容量庞大,如何有效得到目标克隆,是研究者们必须解决的一个难题。目前,从宏基因组文库中筛选新型生物催化剂的策略可分为3种:基于活性的筛选,基于序列的筛选和底物诱导的筛选。
4.1 基于活性的筛选
基于活性的筛选 (Activity-based screening),也称功能驱动筛选 (Function-driven screening),是指通过活性检测手段从基因组文库中获得具有特殊活性的克隆方法。该方法经常借助于显色反应、选择性表型和形成水解圈等特性进行筛选,如 Diaz-Torres等[44]将构建的口腔唾液宏基因组文库涂布于含有四环素的LB平板上,筛选得到了一个四环素抗性基因;本实验室利用生色底物 5-溴-4-3-吲哚-辛酸盐 (5-Bromo-4-chloro-3-indolyl-caprylate,X-cap) 和 5-溴-4-氯-3-吲哚-β-D-半 乳 糖 苷 (5-Bromo-4-chloro-3-indolyl β-D-galactopyranoside,X-gal) 水解后会产生蓝色物质的特性,从土壤宏基因组文库中分别筛选到了多个酯酶基因[18]和 β-半乳糖苷酶基因[26]。目前已利用基于活性的筛选方法获得了许多新型生物催化剂,如木聚糖酶、纤维素酶、酯酶和脂肪酶等[45]。该方法最大的优点是:不依赖于已知序列的信息,故能够筛选到具有优良特性的酶。此外,该方法还可以快速鉴别有开发潜力的克隆子及全长基因,在工业生物催化剂的开发上具有一定的用途。但此方法也存在以下缺点:1) 由于目前DNA文库的构建中所采用的克隆系统是大肠杆菌系统,真核生物的基因在这个系统中往往无法表达,因而提取到的宏基因组中存在的真核生物基因组DNA会影响到基因组文库的筛选效率;2) 受所选择的表达系统的影响 (宏基因组来源的基因转录效率低;翻译效率低;外源蛋白质难以分泌到胞外;目标蛋白质由于缺乏分子伴侣形成不正确的折叠;缺乏合成的辅因子;与宿主菌密码子使用存在差异),导致许多基因无法正确表达而遗漏;3) 工作量大,筛选效率较低,有时需要分析几千甚至几万个克隆子才能检测到一个阳性克隆子。针对上述不足,研究者们提出了一些改进方法,如在DNA提取过程中,通过差速离心法去除样品中的真核生物,从而减少总DNA中的真核生物DNA[42];应用穿梭载体构建文库,从而使宏基因组DNA能够在大肠杆菌、链霉菌、或者假单胞菌中稳定表达[46-47];也有对大肠杆菌进行修饰以增加基因的表达范围[46]。总而言之,操作时应注意选择合适的载体/宿主系统,优化筛选方法,这样才能提高挖掘新型生物催化剂的效率。
4.2 基于序列的筛选
基于序列的筛选是以序列相似性为基础,根据已知相关功能基因的保守序列设计探针或PCR引物,然后通过杂交或PCR扩增来筛选目标克隆。不断扩大的基因库为这种方法提供了坚实的基础。迄今采用这种方法筛选到的新型催化剂有纤维素酶[11]、铁过氧化物歧化酶[14]、脂肪酶[17],延胡索酸酶[21]、木聚糖酶[32]等。与活性筛选法相比,该方法不依赖于重组基因在外源宿主中表达,且利用基因芯片技术、基因盒系统、荧光原位杂交技术 (Fluorescent in situ hybridization,FISH) 和微阵列技术 (Microarray)还可大大提高筛选效率,并有望获得基因的全长序列。但是该方法仍然存在3个主要的缺陷:1) 由于探针和引物是基于已知序列的保守区域,而大部分具有实际用途的基因在序列上差相很大,通过 PCR扩增或探针杂交很难找到全新的同源基因,因此难以发现新型的酶分子,但有可能筛选到某一类结构或功能的蛋白质中的新分子;2) 一般只能获得结构基因的一个片段,而不是全长基因;3) 对于新鉴定或推测的功能基因需要通过生化测定进一步验证。
4.3 底物诱导的筛选
底物诱导筛选 (Substrate induced gene expression screening,SIGEX) 由Uchiyama等[34]于2005年首次提出,用于筛选那些受底物诱导的靶标基因。Uchiyama等成功的关键在于设计了一个具有操纵子陷阱 (Operon-trap) 的载体 (命名为 p18GFP),该载体含有一个报告基因——绿色荧光蛋白基因 (gfp),并将多克隆位点置于lac启动子和gfp基因之间,使gfp基因的表达受到插入片段中启动子的调控。这样的设计保证了p18GFP载体既能用于构建文库,又能使用荧光激活细胞分离仪 (Fluorescence-activated cell sorting,FACS) 进行高通量筛选[34,48-49]。Uchiyama等采用 p18GFP载体构建了一个含有152 000个克隆子的基因组文库,并对文库进行SIGEX分析:以苯甲酸盐为底物得到58个阳性克隆子;以萘为底物得到4个阳性克隆子。整个筛选过程只用了4 d。由此可见,该方法灵敏度高、快速经济,而且不需要对底物进行修饰;可以从底物推断出目标基因的功能。但 SIGEX法存在以下缺陷:1) 无法利用不能进入细胞质的底物,从而无法诱导相关目的基因的表达。因此,SIGEX法不能用于筛选大分子底物的酶如淀粉酶、蛋白酶、脂肪酶、纤维素酶和木聚糖酶等[51],而这些酶在工业上具有重要用途;2) 对目标基因的结构和插入方向敏感,容易导致漏检,如SIGEX无法检出组成型表达的目标基因,也无法检出与gfp基因插入方向相反的目标基因[50-51];3) 当分解代谢基因和 gfp基因之间存在转录终止子时,阳性克隆子无法被检出,而大的插入片段中通常含有高丰度的转录终止子,所以SIGEX法不能有效筛选大片段插入文库[51];4) 有时仅能获得基因的部分片段[49];5) 对设备要求较高,应用受到一定的限制。
以上3种筛选方法各有优缺点,研究者应根据具体情况进行选择。鉴于它们之间可以相互补充,因此,将它们巧妙结合可以大大提高宏基因组文库的筛选效率。
5 展望
应用宏基因组学挖掘新型生物催化剂是宏基因组学研究中的一个重要方向,引起了全世界科研工作者的关注。但在研究过程中还存在一些亟待解决的问题。首先,现行的环境样品 DNA提取方法还存在很多不足,导致某些环境样品中的基因组DNA无法被有效提取;其次,目前使用的载体/宿主系统使得很多基因 (特别是真核微生物的基因) 无法进行异源表达,无法利用基于活性的筛选及底物诱导筛选两种策略进行筛选;再次,现行的各种筛选方法都存在一些缺陷,无法对宏基因组文库进行全面而有效的筛选;最后,筛选获得的生物催化剂在酶学性质上还存在很多不足,无法应用于工业化生产。根据目前研究中存在的问题,可以从以下几个方面着手:1) 与传统的微生物分离培养技术相结合,使一些不可培养的微生物能够进行富集培养,同时尝试更多的环境样品DNA提取纯化方法,使提取的DNA在产率、纯度、片段长度及种群覆盖率等方面不断提高;2) 探索更加适合构建宏基因组文库的载体和真核表达宿主,同时与宏转录组学结合,挖掘存在于真核微生物中的生物催化剂;3) 与生物信息学结合,设计更加多样、完备的探针和PCR引物,开发更加灵敏、快速、高效的检测手段,提高筛选目标克隆的效率;4) 与蛋白质工程结合,对已获得的生物催化剂进行改造以获得高性能的工业生物催化剂。随着生物学的不断发展,DNA提取方法的突破,更适宜的载体和宿主的获得,文库构建水平和筛选效率的不断提高,宏基因组来源的工业生物催化剂将会不断地被挖掘出来,进而极大地推动工业生物技术的进步。
[1] Torsvik VL. Isolation of bacterial DNA from soil. Soil Biol Biochem, 1980, 12(1): 15−21.
[2] Schmidt TM, DeLong EF, Pace NR. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol, 1991, 173(14): 4371−4378.
[3] Healy FG, Ray RM, Aldrich HC, et al. Direct isolation of functional genes encoding cellulases from the microbial consortia in a thermophilic, anaerobic digester maintained on lignocellulose. Appl Microbiol Biotechnol, 1995, 43(4): 667−674. [4] Stein JL, Marsh TL, Wu KY, et al. Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J Bacteriol, 1996, 178(3): 591−599.
[5] Handelsman J, Rondon MR, Brady SF, et al. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol, 1998, 5(10): R245−R249.
[6] Torsvik V, Øvreås L. Microbial diversity and function in soil: from genes to ecosystems. Curr Opin Microbiol, 2002, 5(3): 240−245.
[7] Fernández-Arrojo L, Guazzaroni ME, López-Cortés N, et al. Metagenomic era for biocatalyst identification. Curr Opin Biotechnol, 2010, 21(6): 725−733.
[8] Courtois S, Cappellano CM, Ball M, et al. Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Appl Environ Microbiol, 2003, 69(1): 49−55.
[9] Yun J, Kang S, Park S, et al. Characterization of a novel amylolytic enzyme encoded by a gene from a soil-derived metagenomic library. Appl Environ Microbiol, 2004, 70(12): 7229−7235.
[10] Park HJ, Jeon JH, Kang SG, et al. Functional expression and refolding of new alkaline esterase, EM2L8 from deep-sea sediment metagenome. Protein Expr Purif, 2007, 52(2): 340−347.
[11] Lee DG, Jeon JH, Jang MK, et al. Screening and characterization of a novel fibrinolytic metalloprotease from a metagenomic library. Biotechnol Lett, 2007, 29(3): 465−472.
[12] Palackal N, Lyon CS, Zaidi S, et al. A multifunctional hybrid glycosyl hydrolase discovered in an uncultured microbial consortium from ruminant gut. Appl Microbiol Biotechnol, 2007, 74(1): 113−124.
[13] Ono A, Miyazaki R, Sota M, et al. Isolation and characterization of naphthalene-catabolic genes and plasmids from oil-contaminated soil by using two cultivation-independent approaches. Appl Microbiol Biotechnol, 2007, 74(2): 501−510.
[14] He YZ, Fan KQ, Jia CJ, et al. Characterization of a hyperthermostable Fe-superoxide dismutase from hot spring. Appl Microbiol Biotechnol, 2007, 75(2): 367−376.
[15] Xu MX, Xiao X, Wang FP. Isolation and characterization of alkane hydroxylases from a metagenomic library of Pacific deep-sea sediment. Extremophiles, 2008, 12(2): 255−262.
[16] Mayumi D, Akutsu-Shigeno Y, Uchiyama H, et al. Identification and characterization of novel Poly (DL-lactic acid) depolymerases from metagenome. Appl Microbiol Biotechnol, 2008, 79(5): 743−750. [17] Tirawongsaroj P, Sriprang R, Harnpicharnchai P, et al. Novel thermophilic and thermostable lipolytic enzymes from a Thailand hot spring metagenomic library. J Biotechnol, 2008, 133(1): 42−49.
[18] Li G, Wang K, Liu YH. Molecular cloning and characterization of a novel pyrethroid-hydrolyzing esterase originating from the Metagenome. Microb Cell Fact, 2008, 7(1): 38.
[19] Rashamuse K, Magomani V, Ronneburg T, et al. A novel family VIII carboxylesterase derived from a leachate metagenome library exhibits promiscuous β-lactamase activity on nitrocefin. Appl Microbiol Biotechnol, 2009, 83(3): 491−500.
[20] Zhao SG, Wang JQ, Liu KL, et al. Screening and characterization of lipase from a metagenome library of dairy rumen microflora. Chin J Biotech, 2009, 25(6): 869−874.赵圣国, 王加启, 刘开朗, 等. 奶牛瘤胃微生物元基因组文库中脂肪酶的筛选与酶学性质. 生物工程学报, 2009, 25(6): 869−874.
[21] Jiang CJ, Wu LL, Zhao GC, et al. Identification and characterization of a novel fumarase gene by metagenome expression cloning from marine microorganisms. Microb Cell Fact, 2010, 9(1): 91. [22] Zhao SG, Wang JQ, Bu DP, et al. Novel glycoside hydrolases identified by screening a Chinese Holstein dairy cow rumen-derived metagenome library. Appl Environ Microbiol, 2010, 76(19): 6701−6705.
[23] Ghai R, Martin-Cuadrado AB, Molto AG, et al. Metagenome of the Mediterranean deep chlorophyll maximum studied by direct and fosmid library 454 pyrosequencing. ISME J, 2010, 4(9): 1154−1166.
[24] Lee MH, Hong KS, Malhotra S, et al. A new esterase EstD2 isolated from plant rhizosphere soil metagenome. Appl Microbiol Biotechnol, 2010, 88(5): 1125−1134.
[25] Ye M, Li G, Liang WQ, et al. Molecular cloning and characterization of a novel metagenomederived multicopper oxidase with alkaline laccase activity and highly soluble expression. Appl Microbiol Biotechnol, 2010, 87(3): 1023−1031.
[26] Wang K, Li G, Yu SQ, et al. A novel metagenome-derived β-galactosidase: gene cloning, overexpression, purification and characterization. Appl Microbiol Biotechnol, 2010, 88(1): 155−165. [27] Sang SL, Li G, Hu XP, et al. Molecular cloning, overexpression and characterization of a novel feruloyl esterase from a soil metagenomic library. J Mol Microbiol Biotechnol, 2011, 20(4): 196−203.
[28] Yao J, Fan XJ, Lu Y, et al. Isolation and characterization of a novel tannase from a metagenomic library. J Agric Food Chem, 2011, 59(8): 3812−3818.
[29] Yu EY, Kwon MA, Lee M, et al. Isolation and characterization of cold-active family VIII esterases from an arctic soil metagenome. Appl Microbiol Biotechnol, 2011, 90(2): 573−581.
[30] Lim MY, Lee HJ, Kim P. Metagenome resource for D-serine utilization in a DsdA-disrupted Escherichia coli. J Microbiol Biotechnol, 2011, 21(4): 374−378.
[31] Schallmey M, Ly A, Wang CX, et al. Harvesting of novel polyhydroxyalkanaote (PHA) synthase encoding genes from a soil metagenome library using phenotypic screening. FEMS Microbiol Lett, 2011, 321(2): 150−156.
[32] Wang GZ, Luo HY, Wang YR, et al. A novel cold-active xylanase gene from the environmental DNA of goat rumen contents: direct cloning, expression and enzyme characterization. Bioresource Technol, 2011, 102(3): 3330−3336.
[33] Jiang CJ, Li SX, Luo FF, et al. Biochemical characterization of two novel β-glucosidase genes by metagenome expression cloning. Bioresource Technol, 2011, 102(3): 3272−3278.
[34] Uchiyama T, Abe T, Ikemura T, et al. Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat Biotechnol, 2005, 23(1): 88−93.
[35] Ivanova N, Tringe SG, Liolios K, et al. A call for standardized classification of metagenome projects. Environ Microbiol, 2010, 12(7): 1803−1805.
[36] Streit WR, Daniel R, Jaeger KE. Prospecting for biocatalysts and drugs in the genomes of non-cultured microorganisms. Curr Opin Biotechnol, 2004, 15(4): 285−290.
[37] Daniel R. The soil metagenome — a rich resource for the discovery of novel natural products. Curr Opin Biotechnol, 2004, 15(3): 199−204.
[38] Courtois S, Frostegård Å, Göransson P, et al. Quantification of bacterial subgroups in soil: comparison of DNA extracted directly from soil or from cells previously released by density gradient centrifugation. Environ Microbiol, 2001, 3(7): 431−2439.
[39] Zhou J, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Appl Environ Microbiol, 1996, 62(2): 316−322.
[40] Braid MD, Daniels LM, Kitts CL. Removal of PCR inhibitors from soil DNA by chemical flocculation. J Microbiol Meth, 2003, 52(3): 389−393.
[41] Desai C, Madamwar D. Extraction of inhibitor-free metagenomic DNA from polluted sediments, compatible with molecular diversity analysis using adsorption and ion-exchange treatments. Bioresource Technol, 2007, 98(4): 761−768.
[42] Pang H, Feng Y, Tang JL, et al. Construction of cosmid library of metagenome derived from compost sample and the improvement of DNA isolation techniques. Microbiol China, 2009, 36(5): 768−772.庞浩, 封毅, 唐纪良, 等. 堆肥宏基因组柯斯质粒文库的构建和 DNA提取技术的改进. 微生物学通报, 2009, 36(5): 768−772.
[43] Schloss PD, Handelsman J. Biotechnological prospects from metagenomics. Curr Opin Biotechnol, 2003, 14(3): 303−310.
[44] Diaz-Torres ML, McNab R, Spratt DA, et al. Novel tetracycline resistance determinant from the oral metagenome. Antimicrob Agents Chemother, 2003, 47(4): 1430−1432.
[45] Steele HL, Jaeger KE, Daniel R, et al. Advances in recovery of novel biocatalysts from metagenomes. J Mol Microbiol Biotechnol, 2009, 16(1/2), 25−37. [46] Martinez A, Kolvek SJ, Yip CLT, et al. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol, 2004, 70(4): 2452−2463.
[47] Penn J, Li X, Whiting A, et al. Heterologous production of daptomycin in Streptomyces lividans. J Ind Microbiol Biotechnol, 2006, 33(2): 121−128. [48] Uchiyama T, Watanabe K. The SIGEX scheme: high throughput screening of environmental metagenomes for the isolation of novel catabolic genes. Biotechnol Genet Eng Rev, 2007, 24: 107−116.
[49] Uchiyama T, Watanabe K. Substrate-induced gene expression (SIGEX) screening of metagenome libraries. Nat Protoc, 2008, 3(7): 1202−1212.
[50] de Lorenzo V. Problems with metagenomic screening. Nature Biotechnol, 2005, 23(9): 1045−1046.
[51] Yun J, Ryu S. Screening for novel enzymes from metagenome and SIGEX, as a way to improve it. Microb Cell Fact, 2005, 4: 8.
