Effect of Competing Anions on Arsenate Adsorption onto Maghemite Nanoparticles
2012-02-14TuutijrviRepoVahalaSillanpandChen
T. Tuutijärvi*, E. Repo, R. Vahala M. Sillanpää,3 and G. Chen
1 Department of Civil and Environmental Engineering, School of Science and Technology, Aalto University, P.O.Box 15200, FI-00076 Aalto, Finland
2 Laboratory of Green Chemistry, Faculty of Technology, Lappeenranta University of Technology, Patteristonkatu 1,FI-50100 Mikkeli, Finland
3 Laboratory of Applied Environmental Chemistry, Department of Environmental Sciences, University of Eastern Finland, Patteristonkatu 1, FI-50100 Mikkeli, Finland
4 Department of Chemical and Biomolecular Engineering, the Hong Kong University of Science and Technology,Hong Kong, China
1 INTRODUCTION
Arsenic is of environmental concern due to its toxicity and carcinogenicity to living organisms. It can be found in groundwater due to natural occurrence and/or anthropogenic sources. The most noteworthy occurrences of arsenic have been detected in parts of West Bengal and Bangladesh, Taiwan, Northern China,Hungary, Mexico, Chile, Argentina and South-Western United States [1]. Arsenic has both organic and inorganic forms, but in groundwater arsenic exists most commonly as inorganic arsenic in two oxidation states that form oxyanions, arsenate As(V), and arsenite As(III). These forms are more toxic than organic ones[2]. Because of arsenic toxicity, many authorities have set the guideline value to 10 µg·L-1[3-5], which is a challenge for environmental engineers.
Common arsenic removal methods are precipitation/coprecipitation, adsorption, ion-exchange and membrane techniques (reverse osmosis, micro, ultra and nanofiltration) [1, 6]. Among them, adsorption is widely applied. Prior to adsorption, As(III) is often oxidised to As(V) as a pre-treatment in order to enhance the extent of arsenic removal [2]. Conventionally used adsorbents for arsenic removal are activated alumina,different iron compounds (e.g. goethite, hematite,granular ferric hydroxide, ferrihydrite, hydrous ferric oxide and iron oxide coated sand or cement), green sand (KMnO4coated gluconite), activated carbon and copper-zinc granules [2, 6-9]. Adsorption in a single component system is the most often-studied process.Although it does not describe the real conditions, it provides valuable fundamental information about the adsorption process. In arsenic adsorption from real wastewater or contaminated water, the effects of commonly found ions in water should be identified, such as phosphate (), silicate (), sulphate natural organic matter (NOM) [8].
Typical concentration ranges of the aforementioned anions in natural waters are listed in Table 1. Of these anions, phosphate, silicate and NOM could have the most significant effect on arsenate adsorption. The presence of NOM has been observed to compete with arsenic for adsorption sites on hematite [13], ferrihydrite and gibbsite [14, 15] and nano-sized zero valent iron (NZVI) [16, 17], thus inhibiting arsenic adsorption,while the influence of sulphate, nitrate and bicarbonate have been found to be limited or insignificant[2, 6, 7, 10, 18, 19].
Several studies have shown that phosphate and silicate compete with arsenate for binding sites on the iron oxide surface. Both are tetrahedral anions forming oxyanions in water, similar to arsenate. Moreover,phosphate exhibits similar chemical properties to arsenate [12, 20]. Accordingly, the presence of phosphate has resulted in a reduction of arsenate adsorption capacity with ferrihydrite [8], bulk maghemite [2], laterite soil [6], iron hydroxides [10], different soils [21],oxidised sediments [18] and different clay minerals(), nitrate (), bicarbonate () and and soils [22]. A decrease in arsenate adsorption was observed to be strongly pH and phosphate concentrationdependent. Silicate is a major component in many surface and subsurface aquatic systems with different characteristic of oxoanion. It is capable of polymerisation at relatively low solution concentrations and on hydrous oxide surfaces [9]. Arsenate adsorption to iron and aluminium oxides has been found to decrease with an increase in silicate concentration and pH[2, 7, 11, 23].
Nano-sized adsorbents, which include nanomaterials such as nanoparticles and conventional adsorbents covered with a certain nanomaterials, have been investigated as novel material for environmental applications [24]. The advantages of nanoadsorbents include high surface area, low waste formation, their possibility to functionalise with various chemical groups to increase their affinity towards target compounds, and easy separation in the case of magnetic nanoadsorbents. The efficiency of different nanoparticles as adsorbents in water and wastewater treatment has been studied with a variety of contaminants [24, 25].Magnetic nanoparticles, such as jacobsite [26], magnetite [27, 28] and maghemite [29-31], have been successfully applied for arsenic removal.
However, when considering practical applications for arsenic removal in water treatment, it is important to investigate the possible effects of other ions on arsenic adsorption capacity. Until now, only a few studies of such research have been reported with nanoparticles [16, 17, 27], but none with maghemite.The objectives of this research were to study the effect of competing ions on arsenate adsorption by maghemite nanoparticles. Two different maghemite nanoparticles were employed in the study: sol-gel process synthesised maghemite [31] and commercially purchased maghemite. The competing effects of sulphate, nitrate,phosphate and silicate were investigated. Experiments were conducted separately with each ion in an arsenate containing solution. The combined effect of ions and other water characteristics were also observed with a groundwater sample which was spiked with a certain amount of arsenate.

Table 1 Typical concentrations of phosphate, silicate, sulphate, nitrate, bicarbonate and natural organic matter (NOM) in natural waters
① TOC=total organic carbon.
2 EXPERIMENTAL
2.1 Reagents
Stock solutions were prepared with reagent-grade chemicals and double deionised water (15 MΩ·cm,Milli-RX 20, Millipore). Na2SO4was purchased from Merck, NaNO3from J.T. Baker, Na2HPO4from Riedel de Haen, Na2SiO3·9H2O and Na2HAsO4·7H2O from Sigma-Aldrich. HNO3and NaOH used for pH adjustment were obtained from Merck and Riedel de Haen, respectively. Working solutions for experiments were diluted from stock solutions of SO410000 mg·L-1, NO3-N 2000 mg·L-1, PO4-P 100 mg·L-1,SiO21000 mg·L-1and As(V) 1000 mg·L-1. All stock solutions were stored in a cold storage room (+4 °C)either in borosilicate (3NO-) or polypropene bottles. Working solutions applied in experiments were a mixture of As(V) and one competing ion at certain concentrations for study. The details of the maghemite nanoparticles synthesised in the present study can be found elsewhere [31]. The commercial nanopowder was purchased from Sigma-Aldrich. They are denoted as SM (sol-gel maghemite) and CM (commercial maghemite) in the subsequent discussion.
2.2 Batch experiments
The effect of competing ions on arsenate adsorption with SM and CM nanoparticles were studied using batch adsorption experiments with four different competing ions. In addition, a natural groundwater sample spiked with As(V) was also investigated. The initial arsenate concentrations studied were 0.5, 1 and 3 mg·L-1, which could be the realistic amount found in slightly and considerably contaminated groundwater.Four sets of experiments were conducted: (1) sulphate and nitrate, (2) phosphate, (3) silicate and (4) As(V)
spiked groundwater. The blank experiments were run simultaneously with studied As(V) concentrations in double deionised water at pH 3 and 7 with both adsorbents. pH 3 was chosen because it is proven to be the optimal pH for arsenate removal by maghemite nanoparticles [31] and pH 7 is closer to the pH of natural groundwater. Solutions pH was adjusted by 2% HNO3or 0.5 mol·L-1NaOH. The experimental conditions are summarised in Table 2.

Table 2 List of batch experiments conducted with SM and CM nanoparticles to investigate the competing effect of certain anions and groundwater sample on arsenate adsorption (pH and adsorbent used in certain single experiment are expressed with tick mark)
The concentration of competing anions was chosen to represent the range of at least the typical levels found in natural waters (Table 1). Adsorbent mass was 0.015 mg for SM and 0.025 mg for CM and the volume of adsorbate was 150 and 100 ml, respectively[32]. An equilibrium time of 50 h and 200 r·min-1agitation at room temperature (23±2) ºC were applied for all experiments. The concentration of total arsenic was measured by an inductively coupled plasma-mass spectrometer (ICP-MS, Perkin-Elmer Elan 6000,Perkin-Elmer Sciex). Phosphate concentrations were analysed before and after adsorption experiments by flow injection analyser (FIA, Foss FiaStar 5000 analyser) according to European Standard [33]. Arsenate is known to interfere with phosphate analysis by FIA.This interaction was investigated and it was observed to be linear and pH-independent. Silicate samples collected before and after adsorption experiments were analysed by an inductively coupled plasma-optical emission spectrometer (ICP-OES, iCAP 6300, Thermo Electron Corporation).
A groundwater sample was delivered from a well in the municipality of Tuusula, Finland. It was collected in an acid-washed polypropylene bottle and stored in a cold storage room (4 °C) prior to the experiments. The chemical composition of the groundwater used in the tests is summarised in Table 3. The groundwater is suitable for iron-based sorbents, since the concentration of sulphate, nitrate, iron, manganese,chloride, fluoride, silica, phosphate, turbidity and total organic carbon (TOC) meet the water quality criteriafor such an application [34]. The negligible arsenic concentration shows that the well water is safe to consume. It also eliminates the effect of As(III) on adsorption experiments, making the analysis easier.The groundwater sample was spiked with some 0.5 and 1 mg·L-1As(V).

Table 3 Analysis results of groundwater sample used in adsorption tests with sol-gel and commercial maghemite
3 RESULTS AND DISCUSSION
3.1 Sulphate and nitrate
The results shown in Fig. 1 indicate that sulphate and nitrate had an insignificant influence on arsenate adsorption with both adsorbents at studied concentrations. Nitrate adsorbs by outer-sphere complexation[35], thus it would not directly compete for the same adsorption sites with the arsenate (inner-sphere complex). The present results are in agreement with previous studies where nitrate 0-226 mg·L-1(as NO3-N)showed no apparent influence on 0.5 mg·L-1arsenate removal with laterite soil at pH 5.7 [6]. Neither was nitrate inhibition observed with a bulk maghemite at pH 6 with initial 0.2 mg·L-1As(V) and 20 mg·L-1nitrate (as NO3-N) [2]. Sulphate can be adsorbed to form either an outer or inner-sphere complex and the type of bond formed might be dependent on experimental conditions such as sulphate concentrations and the order of anion addition [19]. In previous studies,where sulphate and arsenate were present simultaneously, 300 mg·L-1sulphate did not show significant interference on 0.3 mg·L-1arsenate removal with ferric chloride [23]. Neither did 1000 mg·L-1sulphate compete with 0.5 mg·L-1arsenate on laterite soil [6]nor 250 mg·L-1sulphate with 0.2 mg·L-1arsenate on bulk maghemite [2]. Therefore, the results observed in this study are consistent with literature reports.

Figure 1 The effect of sulphate (as SO4) and nitrate (as NO3-N) on As(V) removal efficiency with sol-gel (SM) and commercial (CM) maghemite at pH 31—As(V) 0.5 mg·L-1; 2—SM: As(V) 0.5 mg·L-1 + sulphate 20 mg·L-1; 3—SM: As(V) 0.5 mg·L-1 + sulphate 250 mg·L-1; 4—SM: As(V) 0.5 mg·L-1 + nitrate 1 mg·L-1; 5—SM: As(V) 0.5 mg·L-1 + nitrate 12 mg·L-1; 6—SM: As(V) 0.5 mg·L-1 + sulphate 20 mg·L-1; 7—SM: As(V) 0.5 mg·L-1 + nitrate 1 mg·L-1
3.2 Phosphate
Table 4 shows that phosphate adsorbs onto both adsorbents in water competing with arsenate ions.Adsorption of studied anions is pH dependent and phosphate adsorption follows the same trend as arsenate. Higher removal efficiencies for arsenate were observed at pH 3, which is observed to be the optimal pH for its adsorption due to a combined effect of arsenate speciation and adsorbent surface charge changes with pH [31]. Figs. 2 and 3 show As(V) adsorption isotherms for SM and CM, respectively, at pH 3 and 7. The final pH values of the solutions were 2.9-3.1 for initial pH of 3 and 6.9-7.1 for initial pH of 7. It is evident from Figs. 2 and 3, and Table 4, that phosphate competes with arsenate at both pH values.This competing effect increases with phosphate concentration and solution pH, independent of the source of the adsorbent.

Figure 2 The effect of phosphate concentration (as PO4-P) on As(V) adsorption on sol-gel maghemite◆ 0 mg·L-1 phosphate; ● 0.5 mg·L-1 phosphate; ▲ 1.5 mg·L-1 phosphate;2.9 mg·L-1 phosphate

Figure 3 The effect of phosphate concentration (as PO4-P) on As(V) adsorption on commercial maghemite◆ 0 mg·L-1 phosphate; ● 0.5 mg·L-1 phosphate; ▲ 1.5 mg·L-1 phosphate;2.9 mg·L-1 phosphate

Table 4 Removal efficiencies of As(V) and phosphate in dual solution with sol-gel and commercial maghemite at pH 3 and 7

Figure 4 Distribution of arsenate (a) and phosphate (b) species as a function of pH [37]
The ability of phosphate to compete with arsenate for maghemite surface sites was expected, since phosphate and arsenate have similar structure and chemical behaviour [20]. Moreover, both are adsorbed as an inner-sphere complex onto iron oxides [36].Phosphate and arsenate are both oxyanions in aqueous solution with three similar speciations which are pH dependent. The distribution graphs for arsenate (graph A) and phosphate (graph B) ions are shown in Fig. 4[37]. Despite phosphate being considered an analogue to arsenate, it has been observed that arsenate is more prominent than phosphate for adsorption on iron(hydr)oxides. The larger size of arsenate ion causes it to interact more strongly with some of the OH groups that remain on the adsorbent surface as noted by study with As adsorption on goethite at pH 2.2 [38]. Furthermore, the constant of the binding affinity value of As(V) at pH 6.8 was found to be seven times greater than that of phosphate on ferric chloride [10].
Despite the fact that arsenate adsorbs more strongly on iron (hydr)oxides than phosphate, the competition with phosphate can occur. Phosphate can compete directly for available surface binding sites with arsenate and indirectly influence adsorption through the alteration of the electrostatic charge at the solid surface [8]. Both direct and indirect effects are influenced by solution pH, anion concentrations, arsenate loading on the adsorbent surface and the bonding type, as well as the surface properties of the adsorbent. One possible explanation for increasing phosphate competition with elevated phosphate concentration (Figs. 2 and 3) could be the decrease in the amount of available surface sites. If the capacity of the adsorbent is sufficient for the adsorption of both ions,competition does not show. Therefore, a diminished amount of surface sites results in the appearance of competition effect. Phosphate effect on electrostatic charge at the solid surface should be negligible, since pH 3 is below pHzpcfor both maghemites (5.7 for SM and 7.5 for CM). An increase in surface loading decreases the pHzpcwith maghemites [32], but not below pH 3. Therefore, the surface charge remains positive.
Further examination of the SM results reveal that the phosphate competing effect increases gradually with elevated As(V) concentration at pH 3, but decreases with increasing As(V) concentration at pH 7.In case of CM, the phosphate effect is similar to SM at pH 7 while at pH 3 it shows the highest As(V) adsorption capacity decrease with 1 mg As(V) per L rather than with 3 mg As(V) per L. This is shown in Table 5,where the decrease of As(V) adsorption capacity is expressed. It is also observed that phosphate competition is higher at pH 7 than 3 and slightly higher with CM than SM. At pH 3 with SM, the increase of initial As(V) concentration results in higher As(V) loading on maghemite surface, thus minimising available surface sites which enhance competition with phosphate.Similar results were observed in another study where arsenate and phosphate competitive sorption was investigated with mixed iron-aluminium oxides as adsorbent [22]. The reason for the different behaviour for CM at pH 3 (Table 5) with initial As(V) 3 mg·L-1is not known, but it could possibly be due to uneven surface properties of CM.

Table 5 The decrease of arsenate adsorption capacity in the presence of phosphate with sol-gel and commercial maghemite at pH 3 and 7
The phosphate competition at pH 7 is higher than at pH 3 with both maghemites. This could be explained by declined capacity due to surface charge. In a previous study, it was noted that the competitive effect of phosphate was more apparent in soils with low adsorption capacity than in soils with high capacity [21]. At pH 7, the surface charge of maghemite is negative with SM and partly negative with CM.Therefore, less adsorption sites are available. The surface area-based maximum As(V) adsorption capacity,qmax(mg·m-2), is three times smaller at pH 7 than 3[31], which also indicates that the adsorption capacity of maghemite is diminished at pH 7.
In contrast with the observation at pH 3, the phosphate competing effect decreases in increasing As(V) concentration at pH 7 (Table 5). Similar behaviour has also been noted in previous research with arsenate adsorption on sediment containing iron and aluminium at pH 7.1 [18]. A possible reason for this behaviour is the small amount of active surface sites,which are negative in charge and change in bonding from bidentate to monodentate in case of arsenate [32].Because of the pH and monodentate bonding arsenate is more freely released to the solution and replaced by phosphate.
3.3 Silicate
Dissolved silicate represents a potentially effective competitive anion for As adsorption, since silicate,like As, forms inner-sphere surface complexes and has an affinity with iron (hydr)oxide surfaces [9]. Table 6 shows that both SM and CM adsorb silicate in water competing with arsenate ions. With both adsorbents, a weak increase in silicate removal efficiency at pH 7 is observed, indicating opposite pH dependence to arsenate adsorption. Soluble silica resembles arsenic acid in structure, but differs in speciation: H4SiO4(pH<9.9),
at still higher pH [39].The reason for slightly higher removal efficiency for silicate at pH 7 is probably due to the formation of dimeric silica species, which occupy a higher percentage of the surface as pH increases [40].
As(V) adsorption isotherms with the presence of silicate at pH 3 and 7 for SM and CM are shown in Figs. 5 and 6, respectively. The final pH of the solutions was 2.9-3.1 at initial pH 3 and 6.6-7.1 at initial pH 7. Both adsorbents show similar behaviour. At starting pH 3 with 10 mg·L-1, silicate did not compete with arsenate, but competition is observed by 30 and 50 mg·L-1silicate with 3 mg·L-1As(V). Whereas at initial pH 7 [Figs. 5 and 6, graph (b)] arsenate adsorption was inhibited by all initial As(V) and silicate concentrations. Slightly higher capacities of arsenate in the presence of 30 mg·L-1silicate compared to 50 mg·L-1silicate values for all As(V) concentrations studied with SM could be explained by the lower final pH values (<7.0). The observed results are consistent with previous studies conducted with iron oxide-based hybrid media [11], oxidised sediments [18] and ferric chloride [23]. The possible explanation for the increase in silicate inhibition with elevated silicate concentration and pH is silica polymerisation, which is a function of both concentration and pH. Polymeric species can coat more of the surface and the adsorption sites on the iron hydroxide than monomeric silica, thus inhibiting As(V) adsorption by steric effects or by decreasing the surface potential [11].

Table 6 Removal efficiencies of As(V) and silicate in dual solution with sol-gel and commercial maghemite at pH 3 and 7
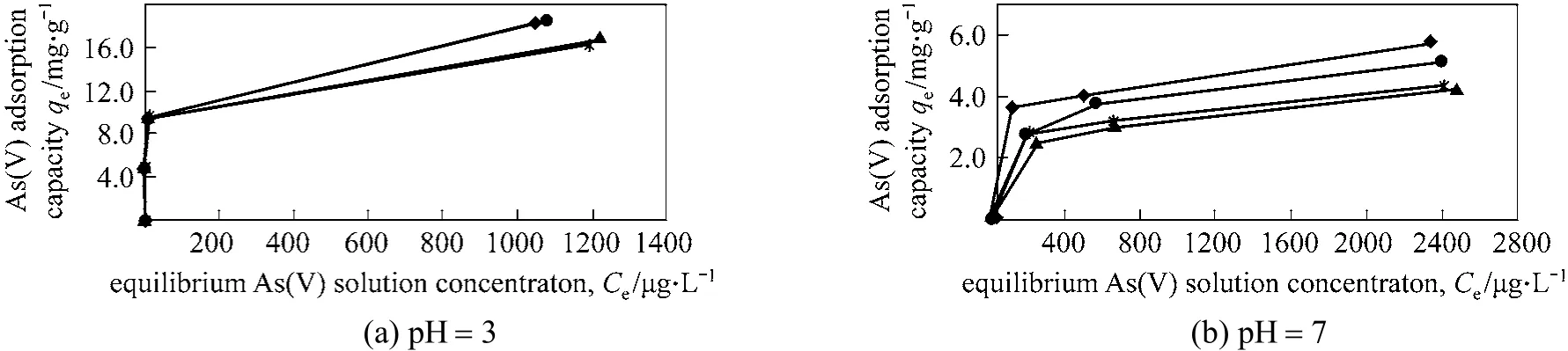
Figure 5 The effect of silicate concentration (as SiO2) on As(V) adsorption on sol-gel maghemite◆ 0 mg·L-1 silicate; ● 10 mg·L-1 silicate; ▲ 30 mg·L-1 silicate;50 mg·L-1 silicate

Figure 6 The effect of silicate concentration (as SiO2) on As(V) adsorption on commercial maghemite◆ 0 mg·L-1 silicate; ● 10 mg·L-1 silicate; ▲ 30 mg·L-1 silicate;50 mg·L-1 silicate
3.4 As(V) spiked groundwater
Table 7 shows the results of adsorption capacities of arsenate from the laboratory and groundwater. The final pH of the solutions was 2.9-3.1 at initial pH 3 and 6.9-7.0 at initial pH 7. The results reveal that bothadsorbents behave similarly. Arsenate adsorption capacity was not affected at studied pH-values with 0.5 mg·L-1As(V) in groundwater, whereas the discrepancy of adsorption results with 1 mg·L-1arsenate concentration is noticeable at pH 3 and 7. At pH 3 the arsenate adsorption capacity decreased by 0.8 and 0.4 units while at pH 7 it increases by 0.8 and 0.6 units with SM and CM when compared with laboratory water spiked with As(V), respectively.
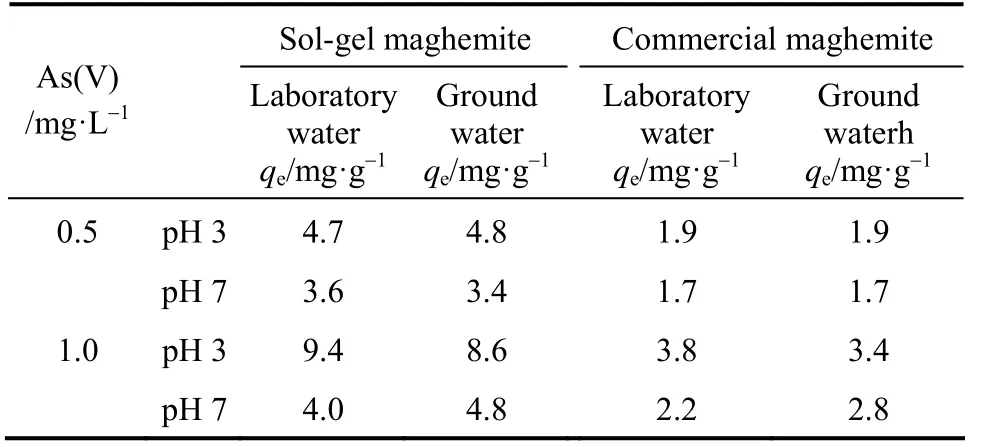
Table 7 Arsenate adsorption capacity with sol-gel and commercial maghemite in laboratory water and groundwater
It has been discussed previously that phosphate and silicate compete with arsenate adsorption. Phosphate influenced strongly on arsenate adsorption capacity with an initial arsenate dose of 0.5 and 1 mg·L-1, even at the lowest PO4-P concentration (0.5 mg·L-1) at pH 7 and a moderate effect was observed at pH 3 (Figs. 2 and 3), while with the same arsenate doses silicate(0-50 mg·L-1SiO2present) was observed to inhibit adsorption only at pH 7 [Figs. 5 and 6, graph (b)].According to the analysis results of groundwater,phosphate and silicate concentrations were <2 μg·L-1and 15 mg·L-1, respectively (Table 3). When comparing these results to separately studied concentrations,in theory, silicate should compete with arsenate at pH 7 with both studied arsenate concentrations (0.5 and 1 mg·L-1). However, this is not in agreement with observed results (Table 7). Phosphate is not considered to show competition with arsenate in the groundwater sample due to its low concentration,which is over 250 times smaller than the lowest separately studied PO4-P concentration (0.5 mg·L-1). At this low concentration, negligible phosphate inhibition is estimated from Figs. 2 and 3 at both pH. It is obvious that the arsenate adsorption results of the groundwater sample cannot be explained by silicate competition and, therefore,the effect of NOM, bicarbonate and ionic strength are further discussed as possible explaining factors.
NOM is a potentially important factor influencing arsenate adsorption onto maghemite nanoparticles.Its effect depends on pH, NOM physicochemical properties (e.g., molecular mass, elemental composition,aromaticity and functional group content) as well as the surface structural properties of the adsorbent [14, 16].NOM consists mainly of high molecular mass humic substances (~80%) such as humic and fulvic acids(HA; FA). NOM molecules have multiple anionic functional groups, which have different dissociation constants and deprotonate at different pH conditions.The majority of them are negatively charged at neutral pH [13]. HA has shown to have a negative surface charge up to pH >2 based on zeta potential measurements [16]. Moreover, a study with FA showed that its effect on arsenate adsorption was highest at pH 4 and decreased at pH 6 and 8 [15]. This was explained by a change in carboxylic groups to phenols at pH>6. Carboxylic groups form inner-sphere while phenols form outer-sphere complexes, the ability of which to compete with arsenate binding sites may be reduced [14, 15].
Based on the aforementioned facts and taking into account the fact that adsorbents surface charge is positive only at pH 3 [32], it is possible that in the present study, NOM had an adverse effect on arsenate removal only at pH 3 with initial As(V) dose of 1 mg·L-1. The effect of NOM with initial As(V) 0.5 mg·L-1was not observed since it is assumed that there is a sufficient amount of active surface sites both for NOM and arsenate. Therefore, competition did not occur. It is supposed that NOM influence on arsenate adsorption at pH 7 decreases due to its negative charge, which has less affinity to negative maghemite surface than arsenate. Moreover, FA forms a possible outer-sphere surface complex at pH 7, which is less favoured for competition with arsenate inner-sphere surface complexes.
Arsenate adsorption capacity increased with initial As(V) concentration of 1 mg·L-1at pH 7. One potential explanation for the result could be a combined effect of bicarbonate and silicate, which has been observed to improve arsenate removal by ~70%when compared to a single silicate effect. The study was conducted with initial As(V) concentration of 0.3 mg·L-1in the presence of (1) 38 mg·L-1silicate and(2) 38 mg·L-1silicate and 134 mg·L-1bicarbonate at pH 6.9 with iron hydroxide as an adsorbent [10]. Another explanation might be the effect of ionic strength.In a previous study it was revealed that increased ionic strength slightly increased arsenate adsorption [initial As(V) concentration 8 mg·L-1] onto maghemite [32].
4 CONCLUSIONS
Arsenate (0.5 mg·L-1) adsorption onto SM and CM nanoparticles was not affected by sulphate (≤250 mg·L-1) or nitrate (≤12 mg·L-1) at pH 3. Instead,phosphate (≤2.9 mg·L-1) was found to be the most prominent anion affecting arsenate (≤3 mg·L-1) adsorption on maghemites. The competing effect was significant both at pH 3 and 7, showing an increase in elevated phosphate concentration and solution pH.The minimal inhibition of phosphate (≤1.5 mg·L-1)on arsenate (0.5 mg·L-1) adsorption was detected at pH 3. Both adsorbents behaved in a similar manner.Phosphate competition with arsenate is possibly influenced by As(V) loading on maghemite surface,phosphate concentration in solution, adsorbent adsorption capacity and surface properties as well as pH.
The competing effect of silicate (≤10 mg·L-1)was negligible for arsenate (≤3 mg·L-1), with both adsorbents at initial pH 3 and noticeable with all concentrations (silicate ≤50 mg·L-1, arsenate ≤3 mg·L-1)at initial pH 7. Slight silicate inhibition was observed with SM (30 and 50 mg·L-1SiO2) and CM (50 mg·L-1SiO2) with 3 mg·L-1As(V) at pH 3. Silicate competition with arsenate is possibly due to its polymerisation,which can inhibit arsenate adsorption by steric effects or by decreasing the surface potential. Silicate behaviour mimicked phosphate, and the competing effect increased with elevated silicate concentration and solution pH.
Arsenate (0.5 mg·L-1) adsorption from groundwater showed almost identical capacities with reference (laboratory water) samples at pH 3 and 7. A discrepancy was observed with 1 mg·L-1As(V). Arsenate adsorption capacity decreased noticeably in the groundwater sample at pH 3 while it increased at pH 7.The capacity decrease at pH 3 is possibly due to NOM competition and capacity increase at pH 7 is possibly a combined effect of silicate and bicarbonate as well as ionic strength effect. Moreover, negatively charged NOM is not disturbing at pH 7 due to negative adsorbent surface, which lowers its attraction towards the maghemite surface. NOM competition was not observed with 0.5 mg·L-1As(V) at pH 3, since there was presumably a sufficient amount of active surface sites to adsorb.
ACKNOWLEDGEMENTS
The technicians at the Civil and Environmental Engineering Department of Aalto University’s School of Engineering are acknowledged for their assistance.We would like to thank Labtium Oy for its professional and fast ICP-MS measurements, and Uusimaa Regional Environment Centre for the collection and delivery of the groundwater sample. The financial support from Maa- ja vesitekniikan tuki ry. is gratefully acknowledged.
1 Nabi, D., Aslam, I., Qazi, I.A., “Evaluation of the adsorption potential of titanium dioxide nanoparticles for arsenic removal”, J. Environ. Sci., 21, 402-408 (2009).
2 Jeong, Y., Fan, M., van Leeuwen, J., Belczyk, J.F., “Effect of competing solutes on arsenic(V) adsorption using iron and aluminum oxides”, J. Environ. Sci., 19, 910-919 (2007).
3 World Health Organization (WHO), Guidelines for Drinking Water Quality, Geneva (2008).
4 European Commission, The Quality of Water Intended for Human Consumption, Council Directive 98/83/EC (1998).
5 U.S. Enviromental Protection Agency (USEPA), “Minor clarification of national primary drinking water regulation for arsenic”, Federal register, 68 (57), 14502-14507 (2003).
6 Maji, S.K., Pal, A., Pal, T., “Arsenic removal from aqueous solutions by adsorption on laterite soil”, J. Env. Sci. Health Pt. A, 42, 453-462(2007).
7 Holm, T.R., “Effects of C O32-/bicarbonate, Si and P O34-on arsenic sorption to HFO”, Jour. AWWA, 94:4, 174-181 (2002).
8 Jain, A., Loeppert, H., “Effect of competing anions on the adsorption of arsenate and arsenite by ferrihydrite”, J. Environ. Qual., 29,1422-1430 (2000).
9 Luxton, T.P., Eick, M.J., Rimstidt, D.J., “The role of silicate in the adsorption/desorption of arsenite on goethite”, Chem. Geol., 252,125-135 (2008).
10 Meng, X, Korfiatis, G.P., Bang, S., Bang, K.W., “Combined effects of anions on arsenic removal by iron hydroxides”, Toxicol. Lett., 133,103-111 (2002).
11 Möller, T., Sylvester, P., “Effect of silica and pH on arsenic uptake by resin/iron oxide hybrid material”, Wat. Res., 42, 1760-1766(2008).
12 Crittenden, J.C., Trussell, R.R., Hand, D.W., Howe, K.J., Tchobanoglous, G., Water Treatment-Principles and Design, 2nd edition,John Wiley & Sons, Inc., New Jersey (2005).
13 Redman, A.D., Macalady, D.L., Ahmann, D., “Natural organic matter affects arsenic speciation and sorption onto hematite”, Environ.Sci. Technol., 36, 2889-2896 (2002).
14 Grafe, M., Eick, M.J., Grossl, P.R., Saunders, A.M., “Adsorption of arsenate and arsenite on ferrihydrite in the presence and absence of dissolved organic carbon”, J. Environ. Qual., 31, 1115-1123 (2002).
15 Simeoni, M.A., Batts, B.D., McRae, C., “Effect of groundwater fulvic acid on the adsorption of arsenate by ferrihydrite and gibbsite”,Appl. Geochem., 18, 1507-1515 (2003).
16 Giasuddin, A.B.M., Kanel, S.R., Choi, H., “Adsorption of humic acid onto nanoscale zerovalent iron and its effect on arsenic removal”, Environ. Sci. Technol., 41, 2022-2027 (2007).
17 Morgada, M.E., Levy, I.K., Salomone, V., Farias, S.S., López, G.,Litter, M.I., “Arsenic(V) removal with nanoparticulate zerovalent iron: Effect of UV light and humic acids”, Catal. Today, 143,261-268 (2009).
18 Stollenwerk, K.G., Breit, G.N., Welch, A.H., Yount, J.C., Whitney,J.W., Foster, A.L., Uddin, M.N., Majumder, R.K., Ahmed, N., “Arsenic attenuation by oxidized aquifer sediments in Bangladesh”, Sci.Total Environ., 379, 133-150 (2007).
19 Wilkie, J.A., Hering, J.G., “Adsorption of arsenic onto hydrous ferric oxide: Effects of adsorbate/adsorbent ratios and co-occurring solutes”, Colloid Surface A, 107, 97-110 (1996).
20 O’Reilly, S.E., Strawn, D.G., Sparks, D.L., “Residence time effects on arsenate adsorption/desorption mechanisms on goethite”, Soil Sci.Soc. Am. J., 65, 67-77 (2001).
21 Smith, E., Naidu, R., Alston, A.M., “Chemistry of inorganic arsenic in soils: II. Effect of phosphorus, sodium and calcium on arsenic sorption”, J. Environ. Qual., 31, 557-563 (2002).
22 Violante, A., Pigna, M., “Competitive sorption of arsenate and phosphate on different clay minerals and soils”, Soil. Sci. Soc. Am. J.,66, 1788-1796 (2002).
23 Meng, X., Bang, S., Korfiatis, G.P., “Effects of silicate, sulfate and carbonate on arsenic removal by ferric chloride”, Wat. Res., 34,1255-1261 (2000).
24 Sharma, Y.C., Srivastava, V., Singh, V.K., Kaul, S.N., Weng, C.H.,“Nano-adsorbents for the removal of metallic pollutants from water and wastewater”, Env. Technol., 30, 583-609 (2009).
25 Savage, N., Diallo, M.S., “Nanomaterials and water purification:Opportunities and challenges”, J. Nanopart. Res., 7, 331-432(2005).
26 Parsons, J.G., Lopez, M.L., Peralta-Videa, J.R., Gardea-Torresdey,J.L., “Determination of arsenic(III) and arsenic(V) binding to microwave assisted hydrothermal synthetically prepared Fe3O4, Mn3O4,MnFe2O4nanoadsorbents”, Microchem. J., 91, 100-106 (2009).
27 Shipley, H.J., Yean, S., Kan, A.T., Tomson, M.B., “Adsorption of arsenic to magnetite nanoparticles: Effect of particle concentration,pH, ionic strength, and temperature”, Environ. Toxicol. Chem., 28,509-515 (2009).
28 Turk, T., Alp, I., Deveci, H., “Adsorption of As(V) from water using nanomagnetite”, J. Env. Eng., 136, 399-404 (2010).
29 Hristovski, K., Baumgardner, A., Westerhoff, P., “Selecting metal oxide nanomaterials for arsenic removal in fixed bed columns: From nanopowders to aggregated nanoparticle media”, J. Hazard. Mat.,147, 265-274 (2007).
30 Park, H., Myung, N.V., Jung, H., Choi, H., “As(V) remediation using electrochemically synthesized maghemite nanoparticles”, J.Nanopart. Res., 11, 1981-1989 (2009).
31 Tuutijärvi, T., Lu, J., Sillanpää, M., Chen, G., “As(V) adsorption on maghemite nanoparticles”, J. Hazard. Mat., 166, 1415-1420 (2009).
32 Tuutijärvi, T., Lu, J., Sillanpää, M., Chen, G., “Adsorption mechanism of arsenate on crystal γ-Fe2O3nanoparticles”, J. Env. Eng., 136,897-905 (2010).
33 International Organization for Standardization, “Water quality. Determination of orthophosphate and total phosphorus contents by flow analysis (FIA and CFA). Part 1: Method by flow injection analysis(FIA)”, SFS-EN ISO 15681-1:2003 (2003).
34 U.S. Enviromental Protection Agency (USEPA), Arsenic Treatment Technology Evaluation Handbook for Small Systems, EPA 816-R-03-014 (2003).
35 Sparks, D.L., Environmental Soil Chemistry, 2nd edition, Academic Press, San Diego, California, USA (2003).
36 Arai, Y., Sparks, D.L., “ATR-FTIR spectroscopic investigation on phosphate adsorption mechanisms at the ferrihydrite-water interface”, J. Colloid Interf. Sci., 241, 317-326 (2001).
37 Saha, B., Bains, R., Greenwood, F., “Physicochemical characterization of granular ferric hydroxide (GFH) for arsenic(V) sorption from water”, Sep. Sci. Technol., 40, 2909-2932 (2005).
38 Lumsdon, D.G., Fraser, A.R., Russell, J.D., Livesey, N.T., “New infrared band assignments for the arsenate ion adsorbed on synthetic goethite (α-FeOOH)”, J. Soil Sci., 35, 381-386 (1984).
39 Iler, R.K., The Chemistry of Silica. Solubility, Polymerization, Colloid and Surface Properties and Biochemistry, John Wiley & Sons,New Jersey, USA (1979).
40 Davis, C.C., Chen, H.W., Edwards, M., “Modeling silica sorption to iron hydroxide”, Environ. Sci. Technol., 36, 582-587 (2002).
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Experimental and Modelling Studies of Biomass Pyrolysis*
- Synergistic Multilayer Adsorption for Low Concentration Dyestuffs by Biomass
- Kinetics of Photocatalytic Degradation of Gaseous Organic Compounds on Modified TiO2/AC Composite Photocatalyst*
- 3D Numerical Study on Compound Heat Transfer Enhancement of Converging-diverging Tubes Equipped with Twin Twisted Tapes*
- Techno-economic Analysis of Distributed Hydrogen Production from Natural Gas
- Error Analysis of Adsorption Isotherm Models for Acid Dyes onto Bamboo Derived Activated Carbon