含FeO熔渣对Zr O2固体电解质侵蚀性研究
2012-01-29高运明王少博黄伟超麦华毅
高运明,王 兵,王少博,黄伟超,麦华毅
(武汉科技大学钢铁冶金及资源利用省部共建教育部重点实验室,湖北武汉,430081)
Zr O2基固体电解质只对氧离子有很强的选择透过性,在冶金中被广泛应用于金属液的定氧、熔渣活度的测定[1-2]、无污染脱氧[2]以及从含氧化物的熔渣[3-5]或熔盐[6-7]中提取Ni、Mg和Ta等金属。在这些电化学反应过程中,Zr O2基固体电解质可能会受到不同性质熔体的侵蚀作用,从而影响到固体电解质的导电特性和使用寿命。熔体对固体电解质材料的侵蚀作用,一般包含有固体电解质材料在熔体中的物理溶解、熔体在固体电解质材料中的渗透或扩散以及固体电解质材料与熔体中有关组分的化学作用。熔体对作为氧离子导体的固体电解质的侵蚀与对传统耐火材料的侵蚀既有相似之处,又有所不同。为维持Zr O2基固体电解质用于可控氧流电解还原[8]提铁时的导电性能,提高其使用寿命,需了解含FeO熔渣对Zr O2基固体电解质侵蚀的影响因素。本文应用热力学计算软件FactSage,模拟计算了静态条件(无电场)下,含FeO熔渣对Zr O2材料的侵蚀随温度、熔渣中FeO含量和渣碱度的变化趋势,并利用Zr O2基固体电解质构建可控氧流电池[8],通过开路与外加电势实验,结合静态模拟计算结果,考察了含FeO酸性渣对Zr O2基固体电解质的侵蚀作用。
1 模拟计算与实验
1.1 熔渣的成分设计
计算选取的母渣成分为:w(SiO2)=58%,w(CaO)=25%,w(Al2O3)=10%,w(MgO)=7%。查阅相图[9]可知,母渣的熔点低于1 300℃。模拟计算和实验中FeO按预设比例配入母渣中。经初步计算并整理,拟定的熔渣成分配比如表1所示。

表1 熔渣成分配比Table 1 Slag compositions of simulation calculation
1.2 模拟计算方法
侵蚀实验往往在净化后的高纯Ar气氛保护下进行,因此,熔渣与Zr O2材料的热力学平衡模拟计算中可不考虑气氛的影响。另外,基于Fact-Sage的计算特点,也不考虑Zr O2材料显微结构的影响。为简化处理,选取100 g渣和100 g MgO稳定的Zr O2(其中含Mg O 2.18 g,Zr O297.82 g)为计算基准体系[10-11],初始输入数据包括CaO、SiO2、Al2O3、MgO、FeO和Zr O2等组元的质量(输入Zr O2时无需考虑其晶型结构),选取FToxid数据库,利用Equilib模块中的纯固相氧化物和压力101 k Pa条件下的Transitions算法进行模拟计算。计算过程中体系总质量保持不变。研究温度或FeO含量变化对材料侵蚀的影响时,选取表1中A1~A5所示的熔渣成分,渣碱度一定;设定温度为1 350~1 600℃,步长为50℃;设定FeO含量为0~40%,步长为10%。研究碱度变化对材料侵蚀的影响时,选取表1中B1~B6所示的熔渣成分,温度固定在1 450℃。
1.3 实验原料与方法
实验原料包括CaO、SiO2、Al2O3、MgO(以上均为分析纯)和FeO,其中FeO由草酸亚铁分解制备。选取表1中A2配比的酸性熔渣(w(FeO)为10%)18 g进行实验,实验装置简图如图1所示。用一端封闭的MgO稳定的Zr O2固体电解质管(简称MSZ,内径7 mm,外径10 mm,长78 mm)作为碳饱和铁液与熔渣之间的隔离膜,构建可控氧流电池。在1 450℃下,进行开路条件下(外电路无电流通过)的侵蚀实验1,以及外加电势电解还原条件下(外电路有电流通过)的侵蚀实验2。实验2在外加4 V电势电解还原前后,还进行了电势扫描,以了解电池导电特性的变化。电解时以纯铁坩埚容器为阴极,以与铁坩埚螺纹连接的铁棒为阴极引线,铁棒外套刚玉保护管;用氧化锆管内的碳饱和铁液作为阳极,钼丝连接石墨棒做电极引线。实验1与实验2的侵蚀总时间都为6.5 h。实验后,取出实验样品,分别切割、磨样,采用Nova Nano SEM400型扫描电镜观察其显微结构和进行能谱分析。
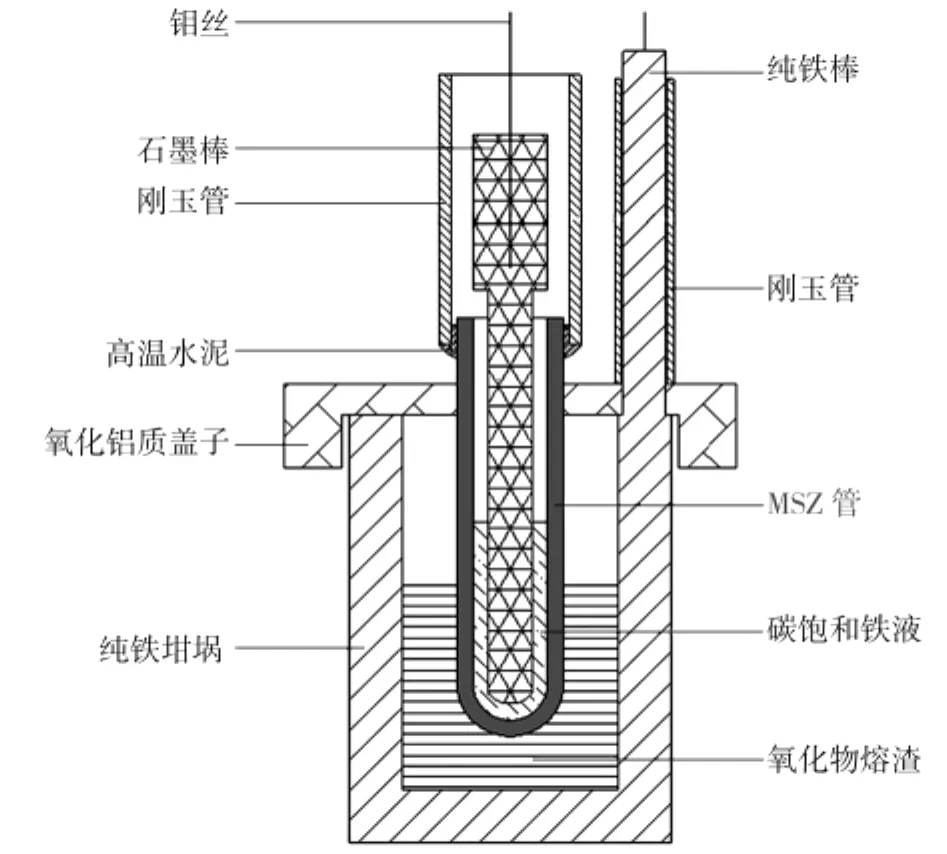
图1 实验装置简图Fig.1 Diagram of experiment device
2 结果与分析
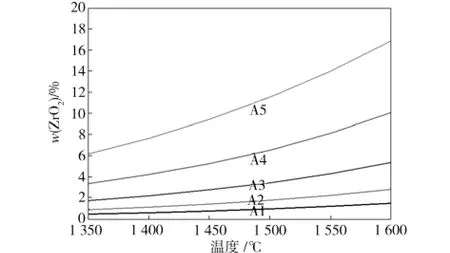
图2 熔渣相中溶解的ZrO2含量变化曲线Fig.2 Curve of ZrO2 content dissolved in slag phase
2.1 热力学模拟计算结果分析
2.1.1 温度及FeO含量对Zr O2材料侵蚀的影响
模拟计算结果显示,当温度在1 350~1 600℃、FeO含量在0~40%范围内变化时,表1中A1~A5渣与Zr O2作用的平衡体系中存在的基本物相为熔渣相和固态四方晶型t-Zr O2相,熔渣相和固态t-Zr O2相中的各物质组成均为氧化物形式。熔渣各组元主要存在于熔渣相,有少量存在于固态t-Zr O2相中;Zr O2组元主要以固态t-Zr O2相存在,少量存在于熔渣相中。熔渣相中除含有A1~A5中的5种基本成分外还含有一定量的Zr O2,固态t-Zr O2相中也含有一定的熔渣成分如CaO、MgO、Al2O3和FeO等。可以认为,熔渣相中含有Zr O2成分只能是由于Zr O2在熔渣中的物理溶解作用造成的,熔渣相中Zr O2含量的大小反映溶解作用的大小;固态Zr O2相中含有熔渣成分是熔渣各组元进入到Zr O2晶界或微孔中造成的,进入的熔渣各组元总含量的多少反映渗透作用的大小。另外,计算结果也显示熔渣与Zr O2之间没有新化合物生成,表明此时熔渣的化学侵蚀作用并不明显。
熔渣相中Zr O2含量随温度和FeO含量变化的趋势如图2所示。由图2中可见,当FeO含量一定时,熔渣相中Zr O2的含量随温度的升高而增大;当温度一定时,熔渣相中的Zr O2含量随FeO含量的增大而显著增大。因此,温度升高以及熔渣中FeO含量的增加均会加剧Zr O2材料在熔渣中的物理溶解,在高温、高FeO含量时尤其显著。
t-Zr O2相中渗透的熔渣各组元的总含量随温度和FeO含量变化的趋势如图3所示。由图3中可见,当FeO含量低于20%时,t-Zr O2相中渗透的熔渣总含量随着温度的升高而略微升高;当FeO含量高于20%时,随着温度的升高,t-Zr O2相中渗透的熔渣各组元总含量反而减小,这可能是由于熔渣相中溶解Zr O2过多导致熔渣各组元在t-Zr O2相中的渗透量下降;当温度一定时,t-Zr O2相中渗透的熔渣组元总含量随着FeO含量的增大而增大。可见,为减小含FeO熔渣对Zr O2材料的侵蚀,应尽可能降低温度,减小熔渣中FeO的含量。考虑到铝电解工业中,冰晶石-氧化铝体系中Al2O3含量一般为2%~5%[12],文献[6]中电解熔盐制取Mg的熔盐中MgO含量也仅为10%,因此在进行可控氧流电解提铁时,熔渣中的FeO含量可以小于20%,在1 350~1 600℃温度区间内,酸性熔渣对氧化锆的侵蚀程度较小,有利于电化学反应的进行。
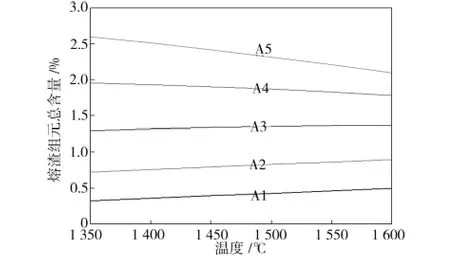
图3 t-ZrO2相中渗透的熔渣各组元总含量变化曲线Fig.3 Curve of slag penetration in t-Zr O2 phase
2.1.2 熔渣碱度对Zr O2材料侵蚀的影响
模拟计算结果显示,1 450℃时,B1~B6渣与Zr O2平衡体系中存在的基本物相有熔渣相、固态t-Zr O2相(含熔渣组元)、固态c-Zr O2相(含熔渣组元)、固态ZrSiO4相和固态CaZr O3相。渣碱度低于0.78时,Zr O2主要以固态t-Zr O2相形式存在;渣碱度高于0.91时,Zr O2主要以固态c-Zr O2相形式存在;渣碱度处于0.78~0.91之间时,固态t-Zr O2相与固态c-Zr O2相共存。各相质量随渣碱度R的变化情况如图4所示。
同时,熔渣相中含有Zr O2成分;t-Zr O2相、c-Zr O2相中含有渣中成分CaO、MgO、FeO、Al2O3等,将其含量分别合并为各相的熔渣组元总含量。熔渣相中Zr O2含量及各相中熔渣组元总含量随渣碱度R变化的情况如图5所示。
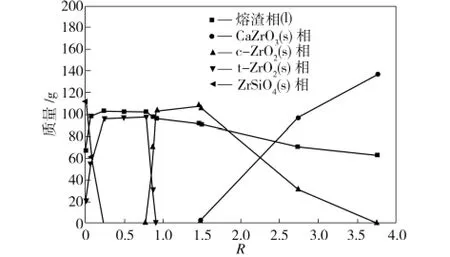
图4 体系中各物相质量随渣碱度的变化Fig.4 Influence of slag basicity on every phase mass changes in system
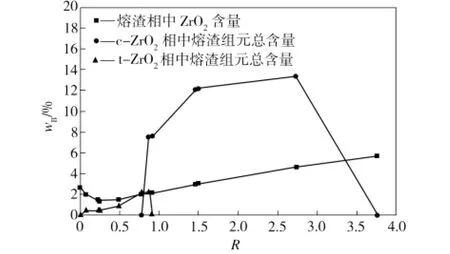
图5 熔渣相和ZrO2相中各物质含量随渣碱度的变化Fig.5 Influence of slag basicity on the relative content of phases in slag phase and ZrO2 phase
由图4和图5中可以发现:
(1)0<R≤0.23时,体系中存在的相为熔渣相、四方相t-Zr O2和固态ZrSiO4(新相)。体系总量一定时,随着体系渣碱度的增大,会促进固态ZrSiO4的分解,固态ZrSiO4的质量会逐渐减小到0,而t-Zr O2相和熔渣相质量逐渐增加。总体上Zr O2在熔渣相中的物理溶解量下降,熔渣在t-Zr O2相的渗透作用微弱,Zr O2与熔渣相之间存在化学侵蚀作用。
(2)当0.23<R≤0.78时,体系中主要成分为熔渣相和四方相t-Zr O2,熔渣相总质量与组成随渣碱度变化不大,Zr O2与熔渣相之间没有化学作用,Zr O2的物理溶解量变化不大,但熔渣在四方相t-Zr O2的渗透作用有所增强。
(3)当0.78<R≤0.91时,体系中主要存在熔渣相和四方相t-Zr O2,并出现立方相c-Zr O2,熔渣相总质量变化不大。随渣碱度增大,t-Zr O2相质量减小到0,c-Zr O2相质量增加,直至R=0.91时,t-Zr O2相全部转化为c-Zr O2相,显示出渣碱度对四方相t-Zr O2与立方相c-Zr O2之间转换的促进作用,这可能是由于体系渗透进入Zr O2相中的熔渣成分含有对Zr O2具有掺杂效果的CaO或MgO量增加所致(见图5),CaO或MgO也可能选择性溶解在氧化锆晶粒中[13]。在该碱度范围内,Zr O2材料与熔渣之间没有明显的化学作用,Zr O2在熔渣相中的物理溶解量增加不大,熔渣在四方相t-Zr O2中的渗透作用先稍有增强,后减小到0,但在立方相c-Zr O2中的渗透作用急剧增强。熔渣相中溶解的Zr O2含量以及立方相c-Zr O2中熔渣组元总含量均为3%时,渣碱度约为0.8(如图5所示)。
(4)当0.91<R≤1.46时,体系中存在的物相主要是熔渣相和c-Zr O2相,且随着R的增大,熔渣相总量稍有减小,c-Zr O2相量稍有增加;熔渣相中溶解的Zr O2含量增加,但c-Zr O2相中渗透的熔渣组元总含量增加更为明显,此时c-Zr O2与熔渣相之间没有化学作用。
(5)当1.46<R≤3.80时,体系中存在的主要物相为熔渣相和c-Zr O2相,并出现了新固相CaZr O3。随着R的增大,熔渣相和c-Zr O2相的质量逐渐减少,主要是CaO和c-Zr O2结合生成固态CaZr O3,固态CaZr O3质量逐渐增加,直到c-Zr O2反应完全时,CaZr O3的质量增加到最大值。在该碱度范围内,熔渣对c-Zr O2的侵蚀主要是化学作用,生成新相CaZr O3后材料强度变低,往往导致Zr O2材料的损毁[14]。据文献[14-17]报道,中、高碱度渣(R=1.325,2或4)与部分稳定氧化锆作用可以生成CaZr O3,而低碱度渣(R=0.824,0.511)中则没有CaZr O3生成,与本模拟计算结果基本一致。
因此,在进行可控氧流电解实验时,为降低熔渣对Zr O2固体电解质材料的侵蚀,熔渣最好选择碱度在0.23~0.80之间的酸性渣,此时酸性熔渣对Zr O2的物理溶解和渗透作用均较小,且不会生成ZrSiO4或CaZr O3,还能使Zr O2保持为具有良好导电特性的四方相[18-19]或与立方相共存的状态(共存时碱度约为0.78~0.80)。
2.2 开路条件下实验结果分析
图6为可控氧流电池开路条件下(实验1),熔渣对氧化锆管侵蚀界面的SEM照片。由图6中可见,氧化锆管截面明显分为两层,靠近熔渣的一层含有较多微孔,晶界明显,且微孔或晶界往往渗透进了熔渣,颜色较深,该层可称为渗透层;另一侧较致密,微孔、晶界不明显,其中基本没有熔渣成分,该层可称为原质层。原质层与渗透层存在明显的界限。
图6(b)、图6(c)中各点位的能谱分析结果如表2所示。由表2中可见,图6(b)中1点区域的原质层仍以Mg O稳定的Zr O2存在;图6(b)中的2、3、4点和图6(c)中1、2点区域氧化锆晶界和微孔中存在熔渣成分和溶解的氧化锆成分,这表明在开路条件下,渗透层内存在氧化锆管内部被含FeO熔渣渗透的情况,与模拟计算结果(见图3)一致。按照原质层与渗透层的界限粗略估计,含FeO熔渣对氧化锆管的渗透深度已达到了94%左右,渗透严重。
由表2中还可见,图6(c)中5、6两点区域的熔渣中也存在氧化锆的成分,与图2、图5所示熔渣对氧化锆存在物理溶解作用的模拟计算结果相一致。对比图6(c)中1、2和5、6点的能谱分析结果可以看出,进入氧化锆微孔的熔渣中溶解的氧化锆含量远远大于氧化锆溶解在熔渣本体中的含量。当然,渗透进氧化锆微孔中的熔渣量远小于熔渣本体量。文献[1]利用氧化锆基固体电解质电池测定熔渣中FeO的活度时(开路条件下),尽管氧化锆管浸入熔渣的时间很短(只有3~4 min),也发现含FeO熔渣对氧化锆管的渗透,甚至影响到测定结果。

图6 实验1中氧化锆/熔渣界面的SEM照片Fig.6 SEM images of ZrO2/slag interface under open-circuit condition
在图6(b)和图6(c)中没有发现熔渣与Zr O2之间有新的化学反应物质生成,氧化锆管/熔渣界面平整光滑。本实验用酸性渣A2的碱度R为0.43,前述热力学模拟计算结果已经表明,熔渣在碱度范围0.23<R≤0.78内,不会与Zr O2形成新相。
开路条件下,回路基本无电流通过,但由于实验采用的氧化锆管存在少许电子导电,内部也会通过微弱的电流,导致熔渣中FeO的少量还原(在熔渣/氧化锆界面存在还原出来的微量铁珠如图6(c)中3、4点位所示)。
上述分析表明,可控氧流电池在开路条件下,熔渣对氧化锆固体电解质侵蚀的主要作用是:熔渣沿氧化锆晶界或微孔进入氧化锆管内部的渗透以及熔渣(包括本体和渗透熔渣)对Zr O2的物理溶解。熔渣对Zr O2没有明显的化学侵蚀作用。开路条件下侵蚀实验结果与前述静态条件下的模拟计算结果较一致。
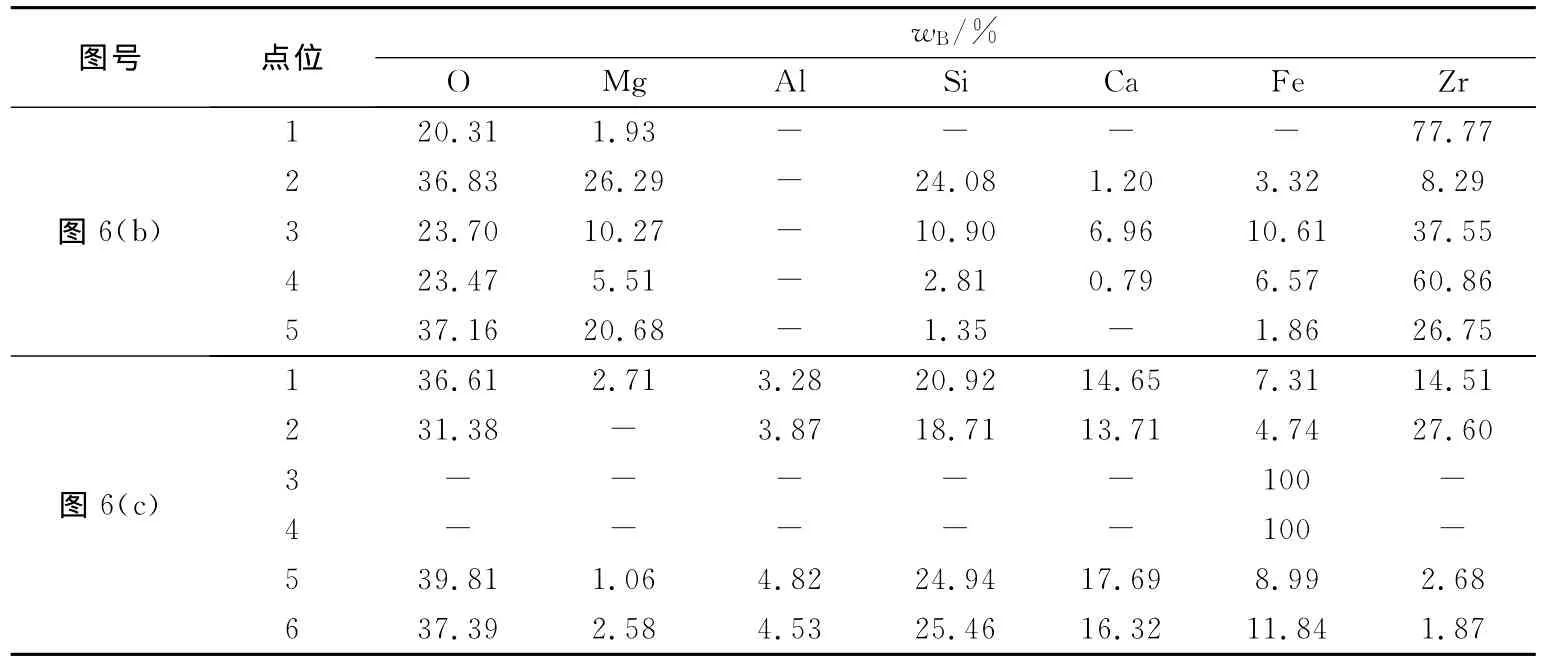
表2 图6中各点位的EDS能谱分析结果Table 2 EDS analysis of point position in Fig.6
2.3 外加电势条件下实验结果分析
图7为可控氧流电池在外加电势电解条件下(实验2)电化学分析仪记录的外电路电流与时间的关系曲线。图8为外加电势条件下铁坩锅/熔渣界面的SEM照片。由图8中可见,电解还原实验后在铁坩埚阴极区证实了有大量金属铁枝晶生成。

图7 外电路电流与时间的关系曲线Fig.7 Curve of external current to time during Experiment 2

图8 铁坩埚/熔渣界面(外加电势条件下)的SEM照片Fig.8 SEM image of Fe crucible/slag interface under applied voltage condition

图9 实验2氧化锆管/熔渣界面的SEM照片Fig.9 SEM image of Zr O2/slag interface under applied voltage condition
外加电势条件下氧化锆管/熔渣界面的SEM照片如图9所示。图9中各点位的EDS分析结果如表3所示。由图9中可见,氧化锆晶粒比较完整,相互接触紧密,熔渣渗透进入氧化锆晶界和微孔中较少,也有氧化锆溶解在熔渣本体(如表3中点位1的数据所示),且在氧化锆管/熔渣界面观察到有ZrSiO4新相层形成。显然,外加电势条件下熔渣对氧化锆管的渗透溶解侵蚀程度要比开路条件下小,但化学侵蚀作用较之更明显。
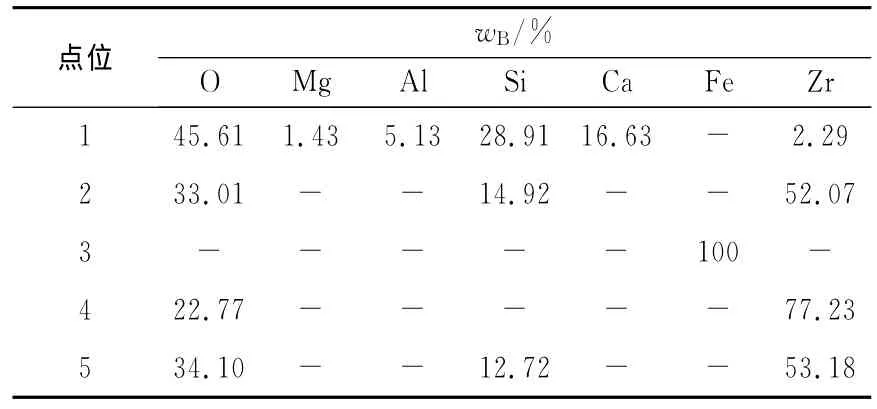
表3 图9中各点位的EDS分析结果Table 3 EDS analysis of point position in Fig.9
由图9中可见,在电解条件下,按照熔渣离子结构理论,熔渣中存在结构简单的Ca2+、Mg2+、Fe2+等阳离子和O2-阴离子以及复杂的硅氧复合阴离子等。在静态以及开路条件下,熔渣中的离子自由散乱分布,在表面张力的推动下,熔渣能沿氧化锆晶界或微孔渗透进入氧化锆管内部,但在外加电势条件下,熔渣中的阳离子向阴极铁坩埚壁面移动,有远离氧化锆管的趋势,且Fe2+在阴极获得电子,首先被还原(见图8):
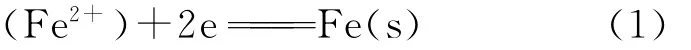
由此导致熔渣中FeO含量不断降低。同时,熔渣中的阴离子O2-向氧化锆管/熔渣界面移动(其移动方向与Zr O2溶解进入到熔渣中离解的O2-迁移方向正好相反,因此能阻碍Zr O2的溶解[20]),进入到氧化锆管内部继续迁移到铁碳熔体侧(阳极),进行氧化反应:
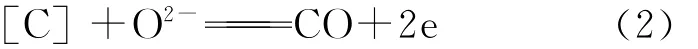
可见,在外加电势作用下,阴、阳离子各自持续向阳、阴极移动,并进行电极反应;外电路有较大的电流通过,熔渣中FeO不断被还原。这些均有利于减小熔渣对Zr O2的溶解作用以及熔渣渗透进入到氧化锆管内部的趋势。另外,由图9中2点和5点位置的成分(参见表3),经过折算,SiO2和Zr O2的平均含量分别为30.6%和69.4%,接近ZrSiO4理论组成(w(SiO2)=32.8%,w(Zr O2)=67.2%),且O、Si、Zr间的原子比接近4∶1∶1,因此,推测其为生成的ZrSiO4新相。5点位置的ZrSiO4应该是由于Zr O2制备过程中原料混入的微量SiO2与Zr O2反应生成的,反应式如下:

式中:ΔGθ为标准吉布斯自由能,J/mol;T为温度,K。
当温度T为1 723 K时,上述反应的ΔGθ=-5 090.2 J/mol<0,表明在标准态下可以生成ZrSiO4。
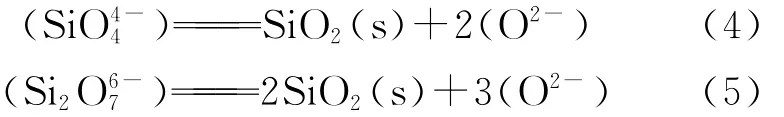
同时,在氧化锆管/熔渣界面会聚集大量的SiO2,甚至从熔渣中饱和析出,导致界面附近熔渣碱度显著降低。若碱度R低于图4所示的理论计算值0.23时,便会发生式(3)反应,在氧化锆管表面生成ZrSiO4。这种化学侵蚀作用会破坏部分稳定Zr O2的晶体结构(表面变得粗糙),增大电池内阻,阻碍氧离子迁移,降低电流效率。图10所示为外加电势条件下电解前后电池的线性扫描伏安曲线,该曲线证实了电解前后电池导电特性的显著变化,而且还原后观察残渣样品,颜色是深绿色的,表明还原并不完全,渣中仍存在相当多的FeO,电池导电特性的显著变化不应该是熔渣中FeO的含量变化所致。当然,高熔点的Zr-SiO4能增大熔渣的黏度,有效减轻或阻止熔渣对氧化锆的渗透溶解。实验中应尽量降低熔渣对氧化锆的化学侵蚀以及熔渣对氧化锆的渗透。为此,结合前述热力学模拟计算结果考虑,适当增大本实验中酸性渣碱度可能是有益的。

图10 实验2电解前后电池的线性扫描伏安曲线Fig.10 Linear scanning current-voltage curves before and after electrolysis of Experiment 2
3 结论
(1)Factsage热力学模拟计算结果表明,在静态条件下(无电场),熔渣对氧化锆材质的物理溶解和渗透程度随温度升高和FeO含量的增大而逐渐加剧;随渣碱度的变化,熔渣与氧化锆之间会有ZrSiO4和CaZr O3生成。
(2)本实验中构建的可控氧流电池在开路条件下,酸性熔渣对氧化锆侵蚀作用主要体现在熔渣沿氧化锆晶界或孔隙进入氧化锆管内部的渗透作用和熔渣本体对氧化锆的物理溶解,没有化学产物新相生成。外加电势条件下,酸性熔渣对氧化锆侵蚀的主要作用是熔渣与氧化锆管之间产生新相ZrSiO4,但同时新相ZrSiO4也显著减轻了熔渣对氧化锆管的渗透和溶解。开路条件下熔渣对氧化锆管的渗透侵蚀比外加电势条件下的严重,但外加电势电解条件下酸性熔渣与氧化锆之间易形成新相,增大电池内阻,阻碍氧离子迁移。
(3)进行可控氧流电解提铁时,为保证固体电解质的固有导电性能,减弱或阻止熔渣对氧化锆固体电解质的侵蚀,熔渣中FeO的含量最好不超过20%,熔渣碱度最好在0.23~0.80之间。
[1] Van Wijngaarden M J U T,Dippenaar R J.The use of zirconia-based solid electrolytes for the rapid determination of iron oxide activities in iron-and steel making slags[J].Journal of the South African Institute of Mining and Metallurgy,1986(11):443-453.
[2] 王常珍.固体电解质和化学传感器[M].北京:冶金工业出版社,2000.
[3] Gao Y M,Guo X M,Chou K C.Pure metal extraction from molten oxide slag by short-circuit galvanic cell[J].Journal of University of Science and Technology Beijing,2004,11(4):306-309.
[4] 高运明,郭兴敏,周国治.短路还原法提取铁的研究[J].金属学报,2006,42(1):87-92.
[5] 孙长余,郭兴敏.非接触式熔融还原法连续制备铁镍合金[J].北京科技大学学报,2010,32(7):900-904.
[6] Krishnan A,Lu X G,Pal U B.Solid oxide membrane process for magnesium production directly from magnesium oxide[J].Metallurgical and Materials Transactions,2005,8(36B):463-473.
[7] Krishnan A,Lu X G,Pal U B.Solid oxide membrane(SOM)technology for environmentally sound production of tantalum metal and alloys from their oxide sources[J].Scandinavian Journal of Metallurgy,2005,34:293-301.
[8] 高运明,姜英,张华,等.可控氧流冶金[J].武汉科技大学学报:自然科学版,2007,30(5):449-453.
[9] 德国钢铁工程师协会.渣图集[M].王俭,彭愉强,毛裕文,译.北京:冶金工业出版社,1989:103.
[10]Berjonneau J,Prigent P,Poirier J.The development of a thermodynamic model for Al2O3-MgO refractory castable corrosion by secondary metallurgy steel ladle slags[J].Ceramics International,2009,35:623-635.
[11]Luz A P,Tomba Martinez A G,Braulio M A L,et al.Thermodynamic evaluation of spinel containing refractory castables corrosion by secondary metallurgy slag[J].Ceramics International,2011,37(4):1 191-1 201.
[12]邱竹贤.有色金属冶金学[M].北京:冶金工业出版社,1995:60.
[13]Suk M O,Park J H.Corrosion behaviors of zirconia refractory by CaO-SiO2-MgO-CaF2slag[J].Journal of the American Ceramic Society,2009,92(3):717-723.
[14]钟耀东,张亚非,强颖怀,等.MgO部分稳定氧化锆陶瓷的抗渣侵蚀性研究[J].耐火材料,2008,42(4):246-249.
[15]Chung Y D,Mark E S.Interaction of CaO-FeOSiO2slags with partially stabilized zirconia[J].Journal of the American Ceramic Society,1994,77(3):611-616.
[16]朱伯铨,李楠,甘芳菲,等.电熔镁锆合成料的抗渣性研究[J].耐火材料,2000,34(3):130-132.
[17]Joo H P.Formation of CaZr O3at the interface between CaO-SiO2-MgO-CaF2(-ZrO2)slags and magnesia refractories:computational and experimental study[J].Computer Coupling of Phase Diagrams and Thermochemistry,2007,31:149-154.
[18]刘继进,何莉萍,陈宗璋,等.氧化钇掺杂四方氧化锆电性能的研究[J].硅酸盐学报,2003,31(3):241-245.
[19]Yamamoto O,Takeda Y,Kanno R,et al.Electrical conductivity of tetragonal stabilized zirconia[J].Journal of Materials Science,1990,25(6):2 805-2 808.
[20]刘常青,唐芳,陈启元,等.析氧阳极固体电解质在冰晶石中的腐蚀行为[J].中南大学学报,2007,38(4):701-705.
[21]梁英教,车荫昌.无机物热力学数据手册[M].沈阳:东北大学出版社,1993.
