柱切换高效液相色谱法分析4种鼠尾草属植物的化学成分
2012-01-25张林东李菲曾灵娜向诚李宝才李鹏
张林东,李菲,曾灵娜,向诚,李宝才*,李鹏,2*
(1.昆明理工大学生命科学与技术学院,云南昆明650224;2.(澳门大学)中药质量研究国家重点实验室,澳门999078)
柱切换(column switching)色谱技术又名色谱-色谱联用技术或多维色谱技术(multi-dimensional chromatography)[1],是在普通的HPLC装置中接入一个切换阀(六通阀或十通阀)实现不同流路之间的相互切换,而达到所需的分离分析目的。柱切换技术具有在线净化、浓缩样品、简化样品预处理等优点,被广泛应用于环境保护、农药残留监测等领域[2],但在中药化学成分分析及定量分析领域应用较少,因此把此技术应用于道地中药的质量控制以及中药材替代品的寻找和质量控制有一定意义。
鼠尾草属Salvia是唇形科Lamiaceae鼠尾草族Salvieae中最大的一个属,全世界约1 050种,广布于热带、亚热带和温带地区,我国大部分地区均有分布[3]。由于该属植物种类较多、分布广、变异大,形态特征相近,种间区分困难,造成其系统分类、品种鉴定及资源利用一直较为混乱[4]。该属多种植物常作为丹参的替代品使用。丹参含有水溶性和脂溶性的两大类有效成分。水溶性成分主要为原儿茶醛、丹参素、丹酚酸等多聚酚酸类成分[5],具有消除自由基、保护胃黏膜[6]等药理作用。脂溶性成分为隐丹参酮、丹参酮IIA等多种菲醌类物质[3],药理作用主要体现在抗菌消炎,抗肿瘤和组织修复等[7-10]上。本实验主要利用双柱切换技术[11]实现对该属植物水溶性和脂溶性成分分别在不同色谱柱上进行定性、定量分析,为鼠尾草属植物中复杂化学成分分析提供一个更好的方法,为鼠尾草植物资源的科学评价和综合利用提供依据。
1 仪器与试药
1.1 仪器Agilent 1200高效液相色谱系统:G1322A脱气机,G1311A四元泵,G1329A自动进样器,G1316A柱温箱,G1315D DAD检测器,预吸附柱Megres-C18色谱柱(4.6 mm×10 mm,5 μm),色谱柱1为Agilent Zorbax Sb-Aq色谱柱(4.6 mm×150 mm,5 μm),色谱柱2为Agilent Eclips XDB-C18色谱柱(4.6 mm×150 mm,5 μm),MXP9960-000二位十通高压切换阀(美国Rheodyne),0.25 μm PEEK管,T2000Y型电子天平(美国双杰兄弟(集团)有限公司),CP-214电子分析天平(梅特勒-托利多仪器(上海)有限公司)。
1.2 试药正品丹参药材购自云南一心堂中药房,其他3种鼠尾草均采自丽江,4种药材经云南省中医中药研究院郭世民研究员鉴定分别为丹参Salvia miltiorrhiza,云南鼠尾草Salvia yunnanensis C.H.Wright,甘西鼠尾草Salvia przewalskii Maxim,栗色鼠尾草Salvia castanea Diels。丹参素、丹参酮IIA、丹参酮I、隐丹参酮、原儿茶醛、原儿茶酸、咖啡酸对照品均购自中国药品生物制品检定所。乙腈(色谱纯,美国Tedia公司),娃哈哈纯净水,其他试剂均为分析纯。
2 实验方法与结果
2.1 样品制备
2.1.1 供试样品溶液的制备将药材充分晒干后,鼓风干燥12 h,粉碎,过60目筛。取生药粉1.0 g,精密称定,精密加入20 mL甲醇,称定质量。超声提取30 min,放冷后称定质量,用甲醇补足损失的质量。摇匀,静置,取上清液,用0.45 μm的微孔滤膜过滤,滤液作为供试样品溶液。
2.1.2 提取工艺加样回收率试验样品制备精密移取一定量的各对照品溶液置于烧杯中,挥干,然后称取1.000 g丹参药材置于烧杯中按照2.1.1项中供试样品的制备方法,提取药材制备提取工艺加样回收率试验样品。
2.1.3 对照品溶液的制备精密称取丹参素6.0 mg,其他对照品均精密称取4.0 mg。丹参素、原儿茶醛、原儿茶酸、咖啡酸分别加到不同10 mL量瓶中甲醇定容。将隐丹参酮、丹参酮I、丹参酮IIA加入到同一个10 mL量瓶中,再从上述定容的4种对照品溶液中分别移取1.00 mL,甲醇定容制成混合标准品贮备液。
2.2 色谱条件柱温为25℃;体积质量1.0 mL/min;0~15 min检测波长(λ)为210 nm,15~70 min为275 nm(根据不同物质的特征吸收波长不同选择),DAD检测器在190~600 nm扫描紫外谱图;流动相为A(0.5%磷酸)-B(乙腈),0~40 min在色谱柱1分析,40~70 min在色谱柱2进行分析。柱切换梯度洗脱程序见表1。
2.3 样品测定采用2.2项中色谱条件进行分析,样品中与对照品对应的色谱峰,通过与对照品色谱图的保留时间和DAD检测器扫描的紫外谱图比对确定。
2.3.1 标准曲线及线性范围取2.1.3项方法配制的混合对照品溶液稀释成丹参素为0.3、0.6、2.4、15、60 μg/mL,原儿茶醛和原儿茶酸均为0.2、0.4、1.6、10、40 μg/mL,咖啡酸为0.8、1.6、10、20、40 μg/mL,隐丹参酮、丹参酮I和丹参酮IIA均为2、4、16、100、400 μg/mL的不同质量浓度的混合对照品溶液,以实验方法中色谱条件进样,以质量浓度C为横坐标,以峰面积A为纵坐标进行线性回归。结果表明,各成分在一定的范围内线性关系良好,见表2。
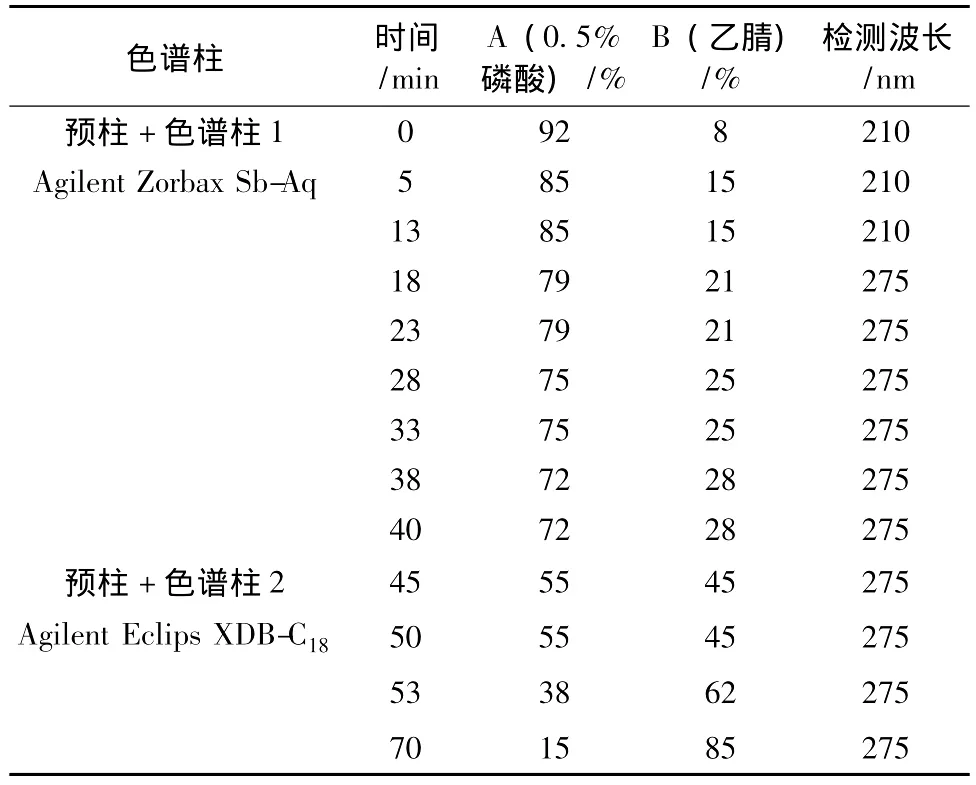
表1 柱切换梯度洗脱程序Tab.1 The gradient program of the column switching HPLC method
2.3.2 最低检测限(LOD)和最低定量限(LQD)取2.1.3项配制的混合对照品溶液稀释成一系列不同质量浓度的对照品溶液,在选定的色谱条件下,按信噪比(S/N)为3和10分别作为最低检测限和最低定量限,结果见表2。
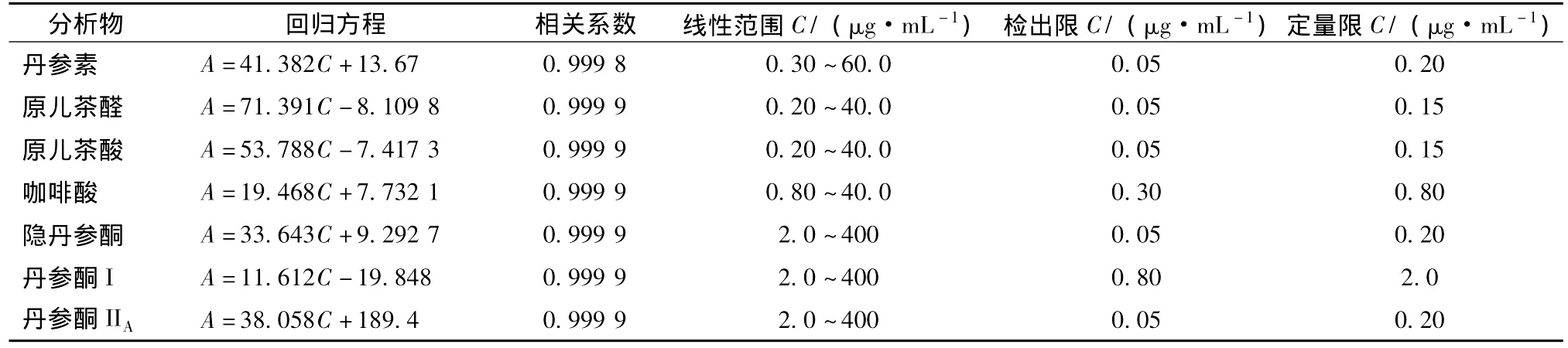
表2 回归方程,相关系数,线性范围,检出限及定量限Tab.2 Regressive equation,correlation coefficient,linear range,LOD and LOQ
2.3.3 提取工艺回收率实验取2.1.2项中制备的丹参加样提取样品分别按照2.2项中色谱条件进行分析,3个添加水平,每个添加水平样品重复进样3次,以3次平均值计算回收率,回收率均在95.23%~98.48%之间,精密度RSD均在0.5%~1.5%之间,见表3。
2.3.4 精密度和回收率实验取2.1.3项中配制的混合对照品贮备液稀释为丹参素15.0 μg/mL,原儿茶醛、原儿茶酸和咖啡酸均为10.0 μg/mL,隐丹参酮、丹参酮I和丹参酮IIA均为100.0 μg/mL的对照品溶液做样品日内、日间精密度,回收率实验。取对照品溶液于同日连续进样5次测定日内精密度,取对照品溶液连续3日,每日进样2次,共6次,测定日间精密度。精密度RSD均在0.12%~0.71%之间,回收率均在95.87%~102.31%之间。
2.3.5 4种鼠尾草属植物中7种成分的测定按照2.1.1项制备供试样品溶液,用建立的柱切换高效液相色谱技术能很好地分离和分析4种鼠尾草属植物中的4种水溶性成分(丹参素、原儿茶醛、原儿茶酸、咖啡酸)和3种脂溶性成分(隐丹参酮、丹参酮I、丹参酮IIA),参照2.3项确定分析物质在色谱图中位置,对照品及甘西鼠尾草提取液的HPLC图谱见图1,7个指标成分的测定结果见表4。
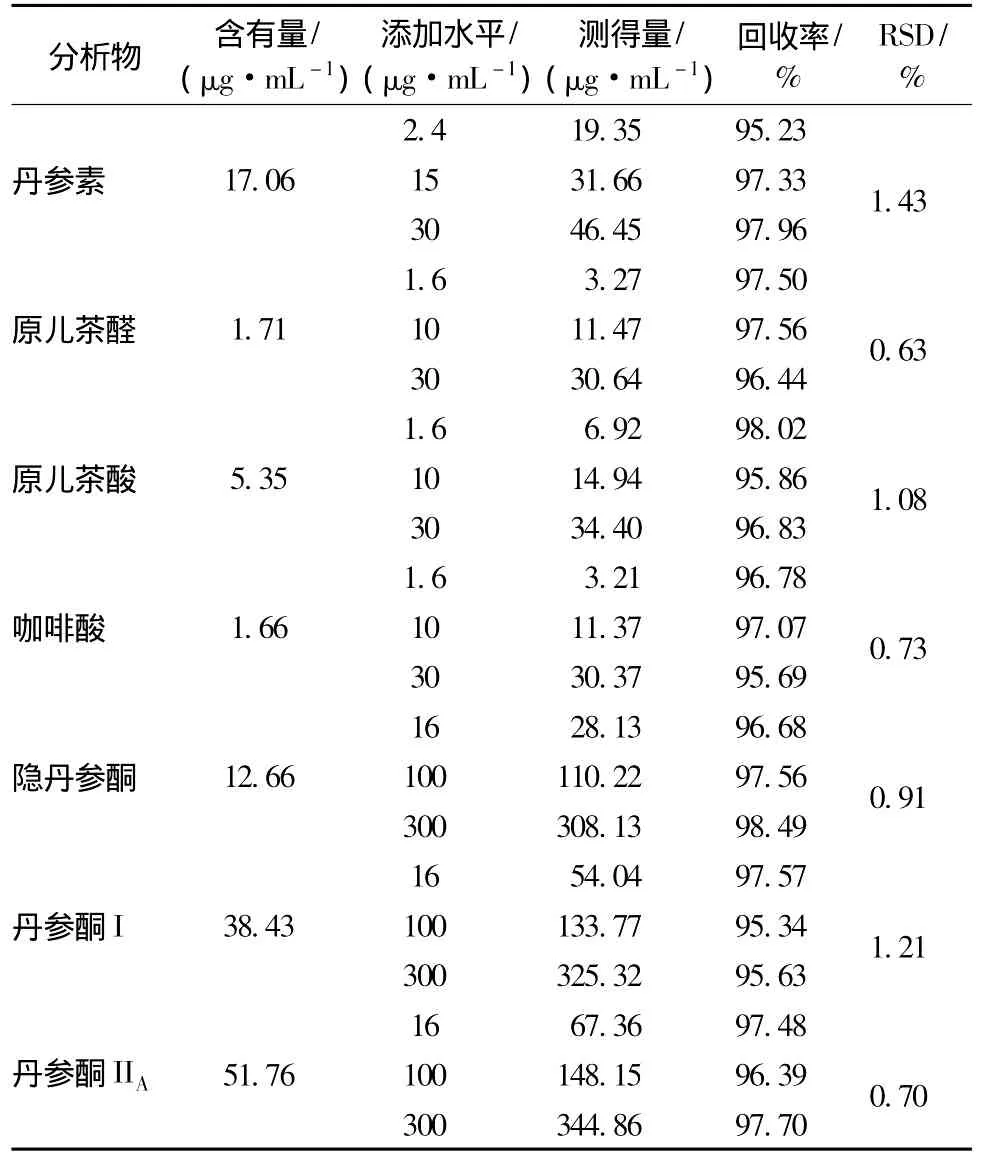
表3 提取工艺加样回收率实验Tab.3 Recoveries of seven components in Savia miltiorrhiza
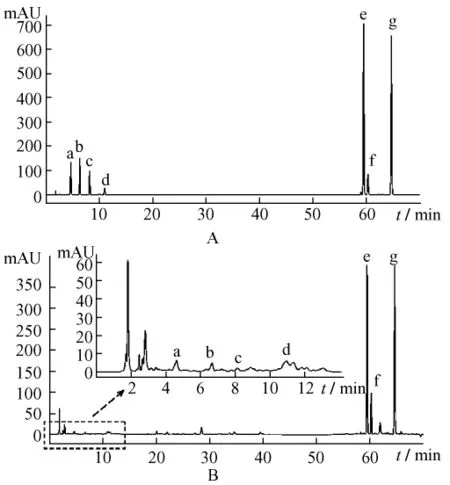
图1 对照品(A)及甘西鼠尾草提取液(B)的HPLC图Fig.1 HPLC chromatograms of referance substances(A)and methanol extract of Salvia przewalskii Maxim(B)
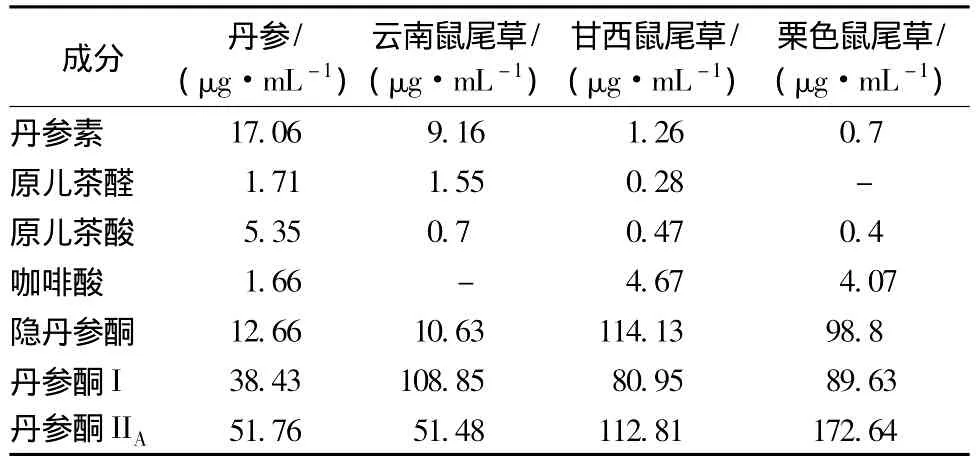
表4 4种鼠尾草属植物中7种化学成分Tab.4 Contents of seven components in four plants of Salvia
3 讨论
3.1 柱切换条件的选择本实验的目的是利用柱切换技术实现更多化学成分的分离,得到分离度好,色谱峰多的色谱图,因此,流动相种类、比例和柱切换时间的选择是重要因素。
3.1.1 流动相的选择为了减弱提取物中酚酸类物质的的拖尾现象和柱切换时系统峰和基线的影响,流动相选择了乙腈-0.5%磷酸水溶液体系。
3.1.2 柱切换时间的选择因为柱切换过程中存在系统死体积,为了使色谱图显现更多的化学成分,切换时间和切换时流动相比例的选择可以避免分析过程中成分的损失,最终确定在40 min,磷酸水溶液比例为28%时进行切换。
3.2 结果分析通过选定色谱条件对4种鼠尾草属植物中7种成分定量结果显示在4种植物中4种水溶性成分除咖啡酸外,含有量均为丹参>云南鼠尾草>甘西鼠尾草>栗色鼠尾草,云南鼠尾草中部分水溶性成分含有量与丹参药材相当,脂溶性成分含有量云南鼠尾草除丹参酮I高于丹参外,另外两组分含有量相当,甘西鼠尾草和栗色鼠尾草三组分均远高于丹参药材,因此得出云南鼠尾草可作为丹参替代药材使用,甘西鼠尾草和栗色鼠尾草在作为丹参药材使用时应选择性的与其他中药材配伍使用。本实验仅对采集样品进行分析。中药材受采集时间,生长环境等影响,其有效成分会存在差异,故本实验结果仅供参考。
[1]Giddings J C.Concepts and comparisions in multidimensional separation[J].J High Resolut Chromatogr,1987,10(5):319-323.
[2]刘海涛,张本刚,陈建民,等.中药中农药残留的分析及其新技术的研究进展[J].中国中药杂志,2006,31(22):1841-1846.
[3]中国科学院中国植物志编辑委员会.中国植物志:第六十六卷[M].北京:科学出版社,1977:70-196.
[4]藤艳芳,王峥涛,余国奠.丹参的药用资源研究进展[J].中国野生植物资源,2003,20(2):1-3.
[5]Zhou L M,Zuo Z,Moses S.Danshen:An overview of its chemistry,pharmacology,pharmacokinetics,and clinical use[J].J Clin Pharmacol,2005,45(12):1345-1359.
[6]张向荣,潘卫三,胡军.丹参对消化性溃疡的研究概况[J].中草药,2000,31(8):附11-附13.
[7]梁勇,羊裔明,袁淑兰.丹参酮药理作用及临床应用研究进展[J].中草药,2000,31(4):304-306.
[8]石乃玉,董华民,黄海金.丹参酮药理及临床应用[J].中国医师杂志,2001,3(2):150-151.
[9]陈向荣,陆京伯,石汉平.丹参的药理作用研究新进展[J].中国医院药学杂志,2001,21(1):44-45.
[10]柳丽,张洪泉.丹参活性成分的现代中药药理研究进展[J].中国野生植物资源,2003,22(6):1-4.
[11]Qian Z M,Wan J B,Zhang Q W,et al.Simultaneous determination of nucleobases,nucleosides and saponins in Panax notoginseng using multiple columns high performance liquid chromatography[J].J Pharmaceut Biomed Anal,2008,48(5):1361-1367.
