农吉利有效成分的HPLC指纹图谱研究
2012-01-25张薇樊轻亚范华均黄晓文佘旭辉刘小琴
张薇,樊轻亚,范华均,黄晓文,佘旭辉,刘小琴
(1.广东药学院基础学院,广东广州510006;2.广东药学院药科学院,广东广州510006;3.信阳职业技术学院药学与检验系,河南信阳464000)
农吉利系豆科猪屎豆属植物野百合Crotalaria sessiliflora L.的干燥全草,也称鼠蛋草、响铃草、芝麻响铃铃、细叶芝麻铃,其味淡、性平,有解毒、抗癌的功能,主要含有生物碱和黄酮类化合物。近年来有关农吉利及其有效成分的研究逐渐增多[1-3]。农吉利主要分布于亚洲东南部和日本等地,各地药材的质量及其差异的相关研究还未曾见有报道。本实验考虑到农吉利中黄酮和生物碱两类化合物在有效成分的含有量、性质及色谱分离行为上存在较大差异,通过样品前处理,采用液相色谱-电喷雾-串联质谱(LC-ESI-MS/MS)法鉴定其主要成分,以牡荆素和农吉利甲素分别作为参照峰,利用高效液相色谱(HPLC)法建立了黄酮和生物碱两类化合物有效成分的HPLC指纹图谱。考察了10批农吉利药材,同时定量测定了牡荆素、农吉利甲素,为该药材的鉴别以及其质量评价提供依据。
1 仪器和材料
1.1 主要仪器与试剂LC—10液相色谱仪(带LC-Solution色谱工作站,日本岛津公司);Aglient 1100 HPLC—MS Trap XCT(美国Agilent公司);Acquity UPLC—Q—Tof Micro联用仪(美国Waters公司);PJ21C-AN微波炉(美的公司);HH—6恒温水浴锅(江苏金坛宏华仪器厂);RE52CS—1旋转蒸发器(上海亚荣生化仪器厂);pHS—25数显pH计(上海精密科学仪器有限公司)。
牡荆素对照品(中国药品生物制品检定所,批号:111687-200701);异牡荆素对照品(上海永恒生物科技公司,批号:38953-85-4);农吉利甲素对照品(美国New Jersey公司,批号:315-22-0);甲醇,乙腈(色谱纯,Merk公司);除注明外其他试剂均为分析纯;试验用水为超纯水。
指纹图谱分析软件为国家药典委员会的中药色谱指纹图谱相似度评价系统(2004A版)。
1.2 试药从市场购买不同产地的农吉利样品共10份,药材样品来源见表1,经本校中药学院刘基柱副教授鉴定均为豆科植物野百合的干燥带叶茎枝。
2 方法与结果
2.1 溶液的制备
2.1.1 对照品溶液的制备准确称取牡荆素、异牡荆素和农吉利甲素对照品各5.0 mg,用甲醇溶解后定容至1 0 mL,分别配制成质量浓度为0.50 mg/mL贮备液,保存于冰箱。使用时按需要逐级稀释。
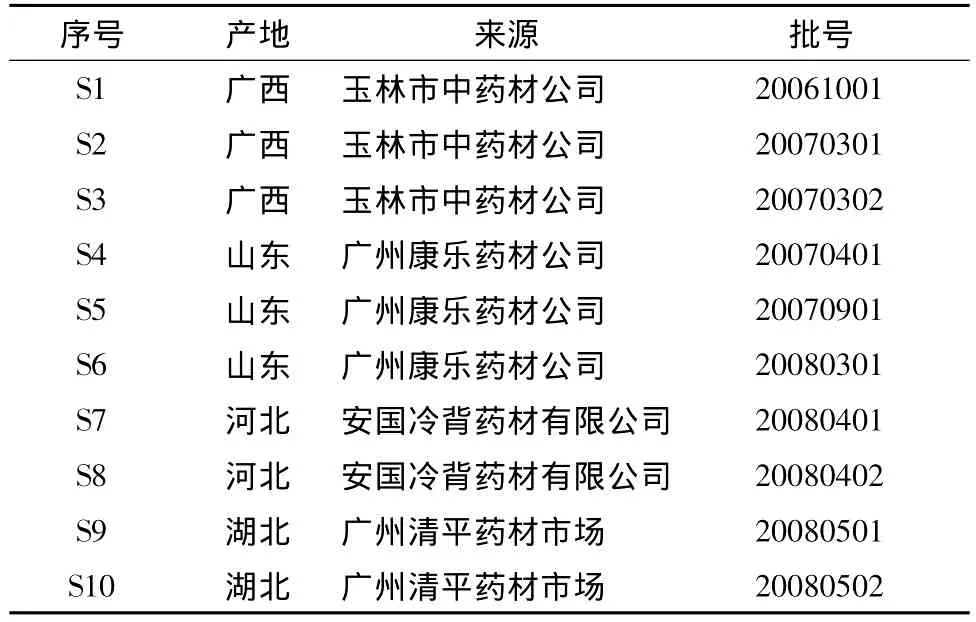
表1 农吉利药材来源Tab.1 Samples of Crotalaria sessiliflora L.
2.1.2 供试品溶液的制备[4-6]称取80目的药材粉末5.0 g于烧瓶中,加入150 mL甲醇,置于微波炉中,调节微波功率780 W,分别加热回流50 min和100 min后,旋干,得到浸膏。平行制备两份。
黄酮化合物供试品溶液的制备一份加入60 mL水溶解,再将其通过AB-8大孔吸附树脂柱,用水洗涤后,吸附的黄酮化合物再用90%乙醇(V/V)洗脱,旋干洗脱液,用甲醇溶解,定容至10 mL。
生物碱供试品溶液的制备另一份加入0.1 mol/L HCl溶液50 mL溶解,转移至分液漏斗中,加入20 mL三氯甲烷萃取,弃去下层,重复3次;再滴加氨水,调节上层溶液酸度至pH 10~11,然后用20 mL三氯甲烷萃取3次,合并三氯甲烷层,旋干溶剂后用甲醇溶解,并定容至5 mL。
2.2 色谱分析
2.2.1 HPLC分析条件黄酮化合物和生物碱的供试品溶液分别以甲醇(A)-0.5%冰乙酸溶液(B)和甲醇(C)-0.04%氨水溶液(D)为流动相,采用梯度洗脱程序分离(见表2);UltimateTMC18色谱柱(4.6 mm×250 mm,5 μm);DAD检测器;柱温30℃;体积流量1.0 mL/min;进样量10 μL。

表2 HPLC梯度洗脱程序Tab.2 Procedures of gradient elution for flavonoids and alkaloids by HPLC
2.2.2 LC-MS分析条件
2.2.2.1 黄酮化合物的分析条件紫外检测器,检测波长330 nm;其他条件同2.2项下黄酮化合物的色谱分离条件。
电喷雾离子阱多级质谱仪ESI离子源;正离子和负离子检出模式;离子源温度110℃;质量扫描范围50~600 m/z;干燥气(N2)体积流量600 L/h;干燥气温度350℃;毛细管电压3.5 kV;雾化气压力50 psi(1 psi=6.895 kPa)。
2.2.2.2 生物碱的分析条件Aquity UPLC BHE C18色谱柱(2.1 mm×50 mm,1.7 μm);流动相为乙腈(E)-0.5%乙酸胺水溶液(F),采用梯度洗脱程序(0~3 min,5%E;3~15 min,5%~30%E;15~17 min,30%~50%E;17~20 min,50%E;20~21 min,50%~95%E);紫外检测波长216 nm;柱温30℃;体积流量0.3 mL/min;进样量5 μL。
电喷雾Q-TOF质谱仪ESI离子源;正离子检出模式;离子源温度100℃;质量扫描范围10~600 m/z;气化室温度350℃;雾化气(N2)体积流量60 L/h;脱溶剂气(N2)体积流量600 L/h;毛细管电压3.5 kV;锥孔电压15 eV。
2.3 方法学的考察
2.3.1 精密度、重复性、稳定性考察根据多批药材的色谱图谱的初步分析结果,取如图1、图2所示的9种黄酮化合物和4种生物碱组分作为共有峰,通过取同一批药材按实验方法制备供试品溶液,用HPLC测定,考察了方法的精密度、重复性(取6份药材)和稳定性试验(分别在0、4、8、12、24和48 h测定)。

图1 农吉利药材黄酮化合物色谱图Fig.1 HPLC chromatogram of flavonoids in Crotalaria sessiliflora L.

图2 农吉利药材生物碱的色谱图Fig.2 HPLC chromatogram of alkaloids in Crotalaria sessiliflora L
黄酮化合物9种组分的色谱峰峰面积的精密度、重复性、稳定性的RSD分别为0.20%~1.7%,0.11%~1.19%,0.23%~1.4%,保留时间的RSD在0.53%~1.1%之间;4种生物碱组分的色谱峰峰面积精密度、重复性、稳定性的RSD分别为0.19%~2.2%,0.39%~2.4%,0.22%~2.8%,保留时间的RSD在0.47%~2.1%之间。结果表明农吉利的黄酮化合物和生物碱组分的保留时间稳定,方法的精密度、重复性和稳定性良好,符合指纹图谱中相对标准偏差不得大于3%的规定,因此选定这9个黄酮化合物和4个生物碱组分作为指纹图谱的共有峰。
2.3.2 共有峰的LC-MS鉴别分别取农吉利的黄酮化合物和生物碱的供试品溶液,按照HPLC-MS条件,对色谱图1和图2中的各组分分别采用LCIonTrap-MS和LC-Q-TOF-MS进行了分析,其结果见表3和表4。
根据表3和表4所示的一级质谱和二级质谱信息,黄酮化合物中的峰3和峰4(图1)及生物碱峰1(图2)经与对照品对比,保留时间、紫外光谱、质谱数据均一致,确定为牡荆素、异牡荆素和农吉利甲素(由于农吉利生物碱的稳定性,实验采用Q-TOF-MS更容易获得更精确的分子量信息,有利于结构的鉴别),其他色谱峰通过分析UV光谱、MS及MS/MS谱图数据,并参考相关的文献报道结果[7-14],初步推导了4个黄酮化合物和2个生物碱可能的化学结构。
2.3.3 指纹图谱的建立取10个批次的农吉利药材,分别按2.1.2项制备黄酮化合物和生物碱的供试品溶液,并用HPLC依次测定,分别记录黄酮化合物和生物碱90 min和40 min的色谱图,在10批样品中选择了稳定的9个黄酮化合物和4个生物碱组分作为共有峰,非共有峰占总峰面积均小于10%。鉴于牡荆素和农吉利甲素为农吉利的主要有效成分,且含有量较高、稳定,其色谱峰所占比例大,故分别将其作为黄酮化合物和生物碱图谱的参照峰。采用国家药典委员会开发的中药色谱指纹图谱相似度评价系统研究版(2004A)处理,设定S1样品为参照图谱,将其它样品的色谱峰与参照图谱进行自动匹配,获得农吉利药材指纹图谱共有模式,以及10个批次农吉利药材的指纹图谱和相对峰面积,结果分别见表5、图3。
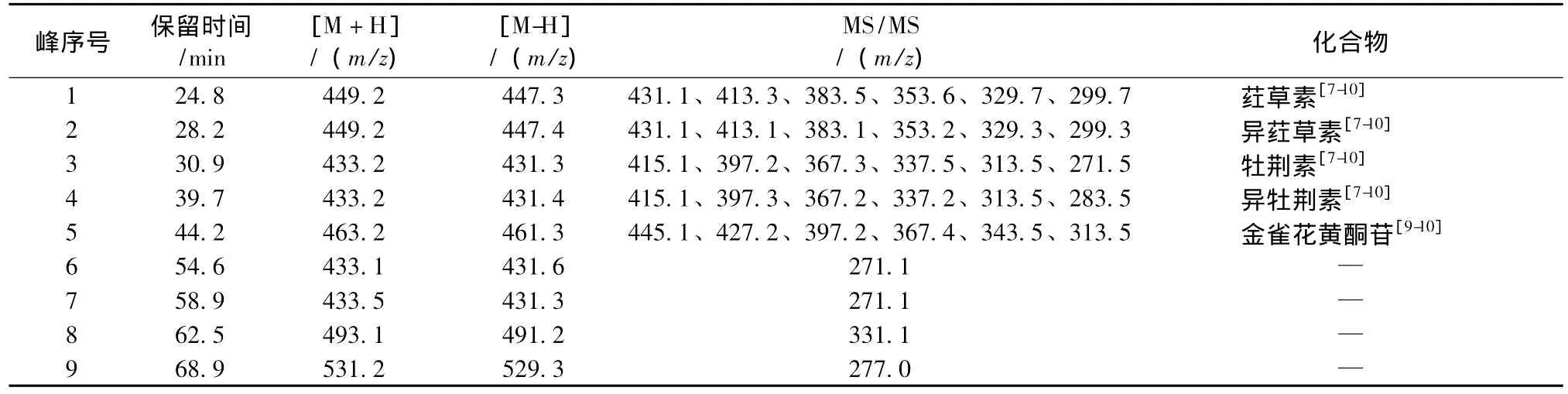
表3 农吉利中黄酮化合物的LC-MS分析结果Tab.3 Obtained data of flavonoids in Crotalaria sessiliflora L.by LC-MS

表4 农吉利中生物碱化合物的LC-MS分析结果Tab.4 Obtained data of alkaloids in Crotalaria sessiliflora L.by LC-MS
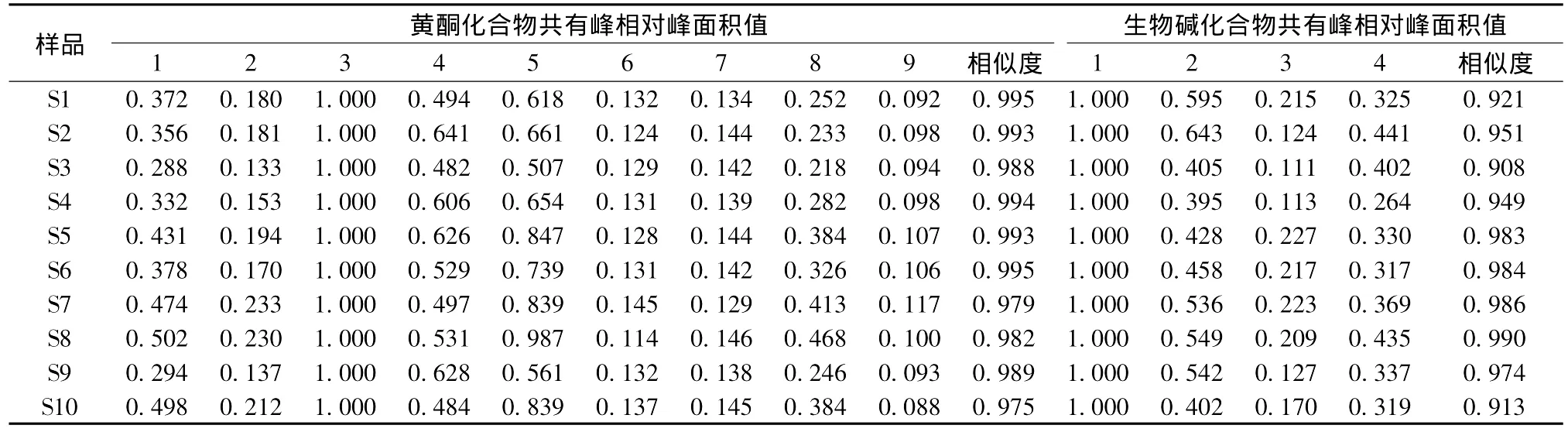
表5 10批药材中黄酮与生物碱化合物共有峰面积及相似度分析结果Tab.5 Relative peak area of common characteristic peaks and similarity analysis for flavonoids and alkaloids in ten batches of samples

图3 农吉利药材中黄酮化合物(A)和生物碱(B)的指纹色谱图Fig.3 HPLC fingerprints of flavonoids(A)and alkaloids(B)in Crotalaria sessiliflora L.
通过计算得出农吉利样品指纹图谱的共有模式,并依此为标准进行比较,各产地的10批农吉利药材相似度均大于0.90,黄酮和生物碱类组分相似度计算结果分别在0.975~0.995和0.908~0.990之间,符合国家药品监督管理局对中药指纹图谱相似度的要求;但不同产地的药材各个组分的色谱峰之间的相对峰面积并不相同,表明药材之间各组分含有量有明显差异,以牡荆素和农吉利甲素含有量最高,作为药材的主要特征有效成分对于药效学、质量标准研究及其开发利用具有重要意义。
2.3.4 主要有效成分测定按照实验方法制备黄酮化合物和生物碱供试品溶液,在文献[5,7]基础上分别测定了10批药材农吉利中主要有效成分牡荆素和农吉利甲素,结果见表6。
表6 不同产地农吉利中牡荆素和农吉利甲素的测定结果±s,n=3)Tab.6 Results of determination of vitexin and monocrotaline in ten batches of samples±s,n=3)
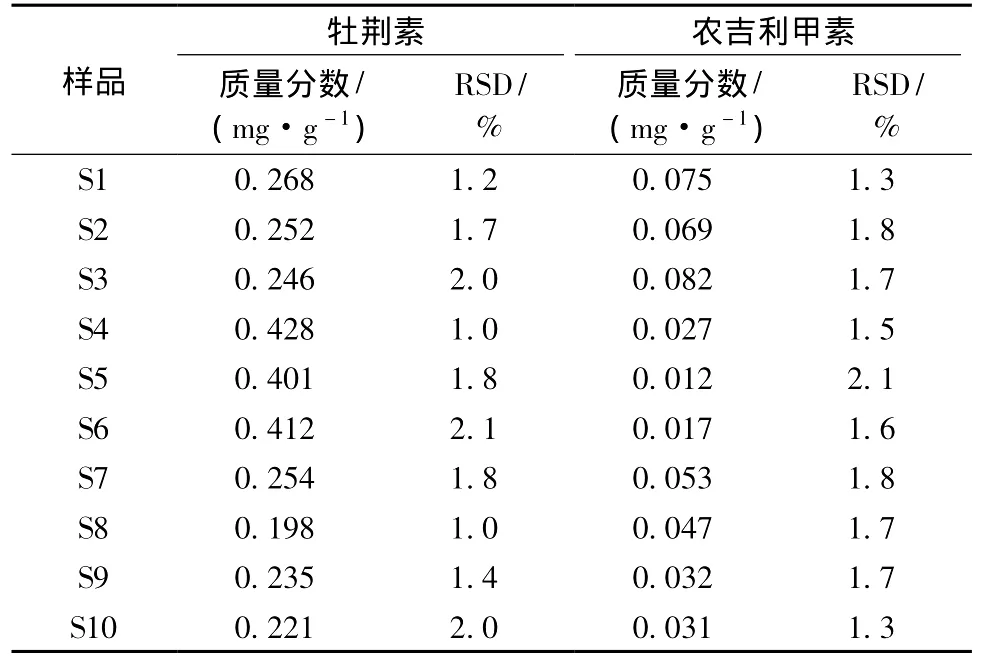
表6 不同产地农吉利中牡荆素和农吉利甲素的测定结果±s,n=3)Tab.6 Results of determination of vitexin and monocrotaline in ten batches of samples±s,n=3)
样品RSD/%S10.2681.20.0751.3 S20.2521.70.0691.8 S30.2462.00.0821.7 S40.4281.00.0271.5 S50.4011.80.0122.1 S60.4122.10.0171.6 S70.2541.80.0531.8 S80.1981.00.0471.7 S90.2351.40.0321.7牡荆素质量分数/(mg·g-1)RSD/%农吉利甲素质量分数/(mg·g-1)S100.2212.00.0311.3
由表6可知,牡荆素和农吉利甲素的质量分数分别在0.221~0.428 mg/g和0.012~0.082 mg/g之间,表明各个产地和批次的农吉利药材含有的牡荆素和农吉利甲素的量有较大差异,其RSD在1.0%~2.1%之间,均小于3.0%,质量稳定,方法的重复性好,精密度高。
3 讨论
3.1 通过实验条件优化,比较了溶剂回流提取、超声提取和微波辐射溶剂回流提取3种方法对药材的提取效果,结果表明,3种方法均含有共同两类有效成分的色谱峰,但微波辐射溶剂回流提取方法的提取效率更高。
3.2 采用甲醇-水、乙腈-水、甲醇-磷酸溶液、乙腈-磷酸溶液、甲醇-乙酸以及甲醇-三乙胺溶液、甲醇-氨水溶液等体系作为流动相分别考察了对黄酮化合物、生物碱成分色谱分离的影响。结果表明:采用甲醇-0.5%乙酸水溶液,在85 min内可完全分离出黄酮提取液中的11个组分,用DAD检测出9种组分在250~280 nm以及300~350 nm之间有两个较强的特征吸收带,与黄酮化合物的紫外特征吸收吻合;采用甲醇-0.04%氨水溶液梯度洗脱,在35 min内可较好地洗脱分离出生物碱提取液中的6个组分,用DAD检测其紫外光谱发现4个组分分别在200~215 nm和215~270 nm有两个吸收谱带,与文献报道的吡咯里西啶生物碱紫外吸收光谱吻合[16]。
3.3 通过对10批不同产地的农吉利药材提取液色谱图的研究,利用HPLC-ESI-MS/MS鉴定和推断了几种黄酮化合物和生物碱成分,以含有量较高的牡荆素和农吉利甲素分别作为黄酮化合物和生物碱指纹图谱的特征参照峰,建立了农吉利中黄酮化合物和生物碱的HPLC指纹图谱分析方法。分别用通过样品前处理后的黄酮化合物和生物碱色谱图建立指纹图谱,各组分分离更简单、纯度更高、指纹图谱的特征性更强,方法的精密度、稳定性、重复性更好,10个产地样品的农吉利黄酮和生物碱的相似度均在0.9以上,符合国家对中药指纹图谱的技术要求。
3.4 通过对不同产地药材指纹图谱的研究和牡荆素和农吉利甲素等组分的峰面积比较,可以发现不同产地的药材基本特征相似,质量稳定,但各个组分的色谱峰面积及含有量不尽相同,尤其是牡荆素和农吉利甲素主要成分的含有量相差较大(分别最大相差近2倍和8倍),如要进一步研究有效成分,控制药材质量和筛选药材,应固定产地及加工方法,以减少因此而导致药材成分的变化。
[1]Prabhakar M C,Bano H,Kumar I,et al.Pharmacological investigations on vitexin[J].Planta Med,1981,43(13):396-403.
[2]Yoo H S,Lee J S,Kim C Y,et al.Flavonoids of Crotalaria sessiliflora[J].Arch Pharm Res,2004,27(5):544-546.
[3]Mun'im A,Negishi O,Ozawa T.Antioxidative compounds from Crotalaria sessiflora[J].Biosci Biotechnol Biochem,2003,67(2):410-414.
[4]樊轻亚,范华均,黄晓文,等.微波辐射/HPLC测定农吉利中牡荆素和异牡荆素的含量[J].中成药,2010,32(2):326-329.
[5]黄晓文,樊轻亚,佘旭辉,等.微波辐射溶剂提取-分光光度法测定农吉利中的总黄酮[J].化学分析计量,2010,19(6):4-6.
[6]张煜帆,樊轻亚,黄晓文,等.微波辐射-溶剂回流提取农吉利中农吉利甲素[J].中国现代应用药学,2009,26(10):819-823.
[7]March R E,Lewrs E G,Stadey C J,et al.A comparison of flavonoid glycosides by electrospray tandem mass spectrometry[J].Int J Mass Spectrom,2006,248(1/2):61-85.
[8]Geoffrey C K,Elaine A P,Fiona C D,et al.Data-directed scan sequence for the general assignment of C-glycosylflavone O-glycosides in plant extracts by liquid chromatography-ion trap mass spectrometry[J].J Chromatogr A,2006,1104(1/2):123-131.
[9]中国科学院上海药物研究所植物化学研究室.黄酮体化合物鉴定手册[M].北京:科学出版社,1981:455.
[10]丛浦珠.质谱学在天然有机化学中的应用[M].北京:科学出版社,1987:421,428.
[11]Kaleab A,Frank S,Michael W.Patterns of pyrrolizidine alkaloids in 12 Ethiopian Crotalaria species[J].Biochem Syst Ecol,2004,32(10):915-930.
[12]Zhou Y,Li N,Choi F F,et al.A new approach for simultaneous screening and quantification of toxic pyrrolizidine alkaloids in some potential pyrrolizidine alkaloid-containing plants by using ultra performance liquid chromatography-tandem quadrupole mass spectrometry[J].Anal Chim Acta,2010,681(1/2):33-40.
[13]Liu F,Wan S Y,Jiang Z J,et al.Determination of pyrrolizidine alkaloids in comfrey by liquid chromatography-electrospray ionization mass spectrometry[J].Talanta,2009,80(2):916-923
[14]赵家俊,方一苇.双吡咯烷类生物碱的特定质谱裂分方向及其特征离子系列的研究[J].分析化学,1981,9(4):418-423.
[15]方一苇,卞则梁,王光辉,等.双吡咯烷类生物碱的质谱断键机理研究[J].化学学报,1981,39(2):139-146.
[16]Betz J M,Eppley R M,Taylor W C,et al.Determination of pyrrolizidine alkaloids in commercial comfrey products(Symphytum sp.)[J].Pharm Sci,1994,83(5):649.
