细胞衰老机制与肿瘤抑制*
2012-01-24沈陈军刘晓平
沈陈军 刘晓平
皖南医学院研究生部,安徽省芜湖市 241002
在自然界中所有具有生命的物体都经历着生老病死,所以衰老(ageing)是生命运动的自然过程,存在于任何生命的任何时期,只是不同情况下其速率与程度有所差别。随着研究的不断深入,分子生物学的不断发展,衰老的研究也从整体水平、器官水平逐渐走向细胞水平、分子水平和基因水平。许多研究发现认为细胞衰老跟人类许多疾病的发病机制有关,如动脉粥样硬化、关节炎、肌肉萎缩退化、溃疡的形成、阿尔茨海默氏病、痴呆、糖尿病、免疫以及癌症。所以根据衰老的诱发因素,可以看出相关的抑癌基因和癌基因的诱导衰老可以抑制肿瘤的发生,但是当衰老功能缺陷时,细胞就会在癌基因刺激下无限增殖成为肿瘤。因此对细胞衰老的研究有助于对肿瘤发生进一步了解,并可能为肿瘤的防治提供方法和线索。
1 细胞衰老的种类
大多数哺乳动物都经历了生老病死有限定的寿命,对此在四十年前Hayflick就已经发现,这一现象是被认为是一种能够防止细胞无限增殖的保护机制,称为细胞衰老(cellular senescence)。目前按衰老的机制不同,将细胞衰老分为增殖衰老和早熟细胞衰老两种类型。
目前将Hayflick发现的由于增殖代数增加引起的衰老称为增殖衰老(replicative senescence),这种衰老通常为细胞生理性衰老,它与细胞随增殖代次增加逐渐丢失端粒末端的序列有关。当端粒缩短到某一临界长度时,端粒功能出现异常,异常的端粒被认为是DNA损伤,从而引发损伤应激反应(DNA damage response,DDR):如p53和Rb等肿瘤抑制基因的激活,使得这些异常的细胞走向衰老或凋亡[1]。除了增殖衰老,近年还发现了早熟细胞衰老(premature cell senescence)现象,即细胞在非端粒信号的刺激下发生衰老,这种形式的细胞衰老与端粒的缩短无关,也与细胞的具体增殖代次无关。细胞早熟衰老最初由Serrano等于1997年在ras基因的研究中揭示,其研究发现与在永生化细胞中不一样,ras在人、鼠原代细胞表达不是促进恶性转化,而是导致细胞周期阻滞,这种现象被称为早熟细胞衰老,这是由DNA损伤、氧化应激、抗肿瘤药物等作用下发生。Braig和Schmitt[2]研究发现细胞衰老还可以在癌基因诱导下发生,被称为癌基因诱导衰老(oncogene induced senescence,OIS),这种衰老被认为是抑制肿瘤形成细胞的自我保护机制之一。Di Micco等进一步证实癌基因诱导衰老是由于癌基因改变DNA的复制进程,激活DNA损伤调控反应所导致,阻断DNA损伤调控反应可以消除癌基因诱导的细胞衰老。
引起细胞衰老的因素有很多,例如由物理以及各种化学等不良因素,自由基累积引起的基因损伤或者由细胞染色体端粒引起的细胞衰老等,是一种复杂的、多因素参与的过程,绝大多数细胞都会出现这种衰老现象。除了物理化学因素和端粒酶功能障碍以外,细胞衰老还可以由癌基因的活化引起。研究表明,将激活的癌基因导入正常细胞可触发防卫反应,阻止细胞增殖。有些致癌基因,如c-Myc一方面可触发细胞凋亡,另一方面可激活ras基因,触发永久的不可逆转的增殖阻滞,导致细胞衰老死亡[3~6]。衰老的研究找到了一些相关的致癌基因,例如普通细胞过表达某些致癌基因(如ras/Raf/MEK/ERK信号连级通路上的一些活性元件)。此外,众所周知几乎所有的人类癌症都缺乏功能基因P53/pRb通路,2007年,Sarkisian等[7]揭示ras诱导细胞衰老与表达量有关,低水平表达可以导致肿瘤发生,而表达量增高则诱导细胞衰老。在体外研究中,这两个关键衰老信号传递路线相互协作,同时也经常携带突变的基因,从而绕过细胞衰老反应引起了肿瘤的发生发展。以上有关细胞衰老的不同概念由于研究者的不同,所以有时早熟衰老又叫加速衰老、应激或异常信号衰老或外在衰老等。所以由此可见细胞诱导领域研究的活跃,对细胞衰老的研究有助于对肿瘤发生进一步了解,并可能为肿瘤的防治提供方法和线索。
2 细胞衰老的相关基因效应通路
细胞衰老的途径被认为是有多重层次的调控,而且这些层次复杂冗余。在此基础上的补充研究认为,至少还有四个衰老基因通路。众所周知细胞衰老和无限增值受信号通路的调控,其中包括对p16 INK4a/pRb通路,p19ARF/p53/p21CIP1/WAF1通路和PTEN/p27KIP1通路等方面的研究进展。其他的基因已经被证实引发衰老,如表型包括PPP1A、SAHH、Csn2、Arase、BRF1、PGM、IGFBP3、IGFBPrP1、PAI-1、MKK3、MKK6、Smurf2和HIC-5。所有的这些基因都已经证实与人的肿瘤有关。然而如前所述所有的这些基因和通路在序列步骤上都遵照一个有序的过程。
两个主要的效应通路可以引起衰老:p14ARF/p53/p21和INK4/CDK/pRb通路[8](如图1)。缺少p53的作用从而诱发显性消极的突变体,特殊的p53反义信使RNA,在细胞培养中寡聚核苷酸或者由病毒引起的癌基因蛋白(例如猿猴病毒T抗原或人乳头瘤病毒16E6蛋白)充分的延长这几个类型细胞的寿命。所以可以认为,衰老和p53功能激活是有关联的[9],也符合端粒缩短、DNA损伤关卡激活和有关的染色体不稳定的结果。在体内研究p53的表现也是一样的[10]。p53的缺失减弱了细胞和有机体的端粒功能障碍的效果,这就说明了p53激活是引起衰老的一个关键的角色作用。
在图1中看到的涉及衰老的其他p53调控的蛋白。根据以往的研究显示,MDM2蛋白能使p53泛素连接酶活化,还能和p53形成一个自动调节相互作用的负反馈回路[11]。MDM2的超表达能够引起p53的降解和其功能丧失。另一个在衰老中基因表达升高的产物就是p14ARF,它能够使被MDM2抑制的p53被重新释放从而恢复活性,并且它能够引起纤维母细胞生长停滞。转基因的鼠胚胎纤维细胞培养诱导产生了ARF蛋白的复合体能够不断的积累,直到细胞进入了衰老。
另一个就是细胞周期蛋白依赖性激酶(cyclin dependent kinase,CDK)抑制剂p21WAF1,它能够直接抑制细胞周期。然而在老鼠的胚胎细胞中,p21WAF1的缺乏却不能够克服细胞的衰老[12]。这就说明至少还有一个额外的下游因子在衰老中需要p53诱导生长停滞。相反的是,在人体细胞中却是不同的,在没有p21的情况下同源复合物的一个双循环有一个足够的能力绕过衰老。一些其他的p53因子可能也在此涉及到,例如14-3-3和GADD45,从而抑制细胞G2和M期的转化,或者myc受体的下调。
视网膜基底部细胞瘤易感基因编码的蛋白pRb也和衰老有关(图1)。过表达的pRb也是一个pRb通路的调节剂和CDK抑制剂,也能模仿衰老表现导致生长停滞。而且通过猿猴病毒大T抗原或人乳头瘤病毒E7癌基因蛋白和腺病毒E1A蛋白诱导pRb的失活,导致细胞寿命的延长[13~15]。
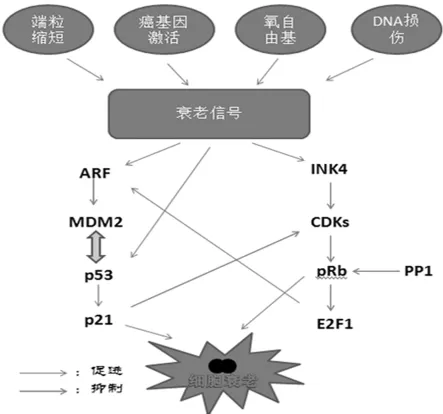
图1 衰老效应通路线路图
已知p16INK4a的功能就是通过CDKs来抑制pRb的失活,p16INK4a功能的减弱被认为很可能就是产生一个类似pRb的功能减弱一样。由此可见在几种人类细胞中p16INK4a积累越多就会越容易衰老。衰老的纤维母细胞可能所含的p16INK4a水平至少是早期传代细胞的40倍。在永生的肿瘤细胞系中p16INK4a的缺失是一样的,并且在体外培养的几个非致癌的永生细胞系中也缺乏p16INK4a蛋白的功能。在野生型的老鼠胚胎纤维母细胞中反转录表达特有的p16INK4a能增加这些细胞永生的几率。和这些表现一致的是,虽然它们显示了正常的衰老效应动力学,但是通过定向敲除鼠细胞p16INK4a基因能够使这些细胞比那些正常受控制的细胞更容易产生永生化。敲除p16INK4a的老鼠能正常的向成年生长并且对生育毫无影响,从而表明个别的INK4蛋白对生长发育并不是必要的。但是p16INK4a的缺乏却产生了对自发性肿瘤发生的低敏感性,同时在对特定的肿瘤治疗方案中能增加这种肿瘤的敏感性。在涉及衰老的通路中,已经发现了一个信号串扰。这个信号串扰很可能就是保证正确的衰老程序功能。而且,在人的原代细胞中例如myc基因所涉及的所有通路中能够绕过衰老。myc通过激活CDK2-细胞周期素A/E复合物从而绕过CDK4/6的抑制并且诱导产生了Cdk激活磷酸酯酶Cdc25A[16]。然而myc又能诱导p27的降解,因此影响了PETN的抑制效果。最后myc的表达通过激活催化亚基的转录来诱导端粒酶的活性[17]。
所有这些步骤,控制这些程序的衰老基因的表达受到DNA甲基化作用的调控。在人类的肿瘤中,在这些基因的启动子区域发生了一个遗传学的改变,使得CpG岛通过异常的DNA甲基化影响了基因的转录调控,使抑癌基因表达发生沉默。在癌症细胞中基因展示的超甲基化有多种途径,包括DNA的修复、细胞循环控制、入侵和转移。超甲基化肿瘤抑制基因BRCA1、p16INK4a、p15INK4b、p14ARF、p73和APC在这中间是沉默不表达的。最近发现S-腺苷高半胱氨酸水解酶(SAHH),在对一个独立的短片断发卡RNA筛选中已经被预先的鉴定出来[18],它的失活能阻碍p53和p16(INK4)诱导的增殖停滞和衰老。SAHH催化S-腺苷高半胱氨酸水解生成腺苷和高半胱氨酸。在真核细胞生物中,这是S-腺苷高半胱氨酸清除的主要路线,S-腺苷甲硫氨酸在其依赖的甲级转移酶作用下失去甲基产生一个共同的产物S-腺苷高半胱氨酸,由此来调节甲基化的进程[19]。有趣的是SAHH的失活抑制了p53的转录控制和削弱了DNA损伤诱导的p21(Cip1)转录。在一份206例患者的不同肿瘤组织和正常组织对比资料显示50%的肿瘤中丢失了SAHH信使RNA。并且SAHH蛋白在对一些结肠癌中也有影响。见图1。
通过以上这些相关通路的具体研究可以发现细胞衰老相关基因之间的关系,INK4a/ARF(inhibitor of cyclin-dependent kianse 4a/alternative reading fram)基因是新发现的一种肿瘤抑制基因(tumor suppressor gene,TSG),p16INK4a和p14ARF分别通过INK4a-CDK4/p16-pRb-E2F1和ARF-MDM2-p53通路履行调控细胞周期的职责。ARF-p53途径的调节主要通过MDM2实现。正常情况下,细胞中的ARF、MDM2和p53水平很低,ARF主要存在核仁中,MDM2和p53则主要在核质中。当有外界信号刺激的时候,可以激活p53表达,p53又可以诱导MDM2基因的转录,而MDM2又具有泛素连接酶E3的作用,可以诱导p53的泛素化,促进p53的降解,降低p53的功能。因此p53和MDM2的相互作用构成了这两种蛋白的负反馈通路(negative feedback loop)。ARF能拮抗MDM2的上述的作用,通过提高p53的功能稳定性或其他尚未知的机制来调节细胞功能。
培养成纤维细胞时在缺乏血清的条件下,当E2F1高表达时能诱导细胞进入细胞周期S期并诱导衰老凋亡。用腺病毒载体介导的E2F1基因转移实验证明,人胃癌、乳腺癌、卵巢癌和结肠癌细胞E2F1过表达可抑制肿瘤细胞生长并诱导衰老和凋亡。E2F诱导凋亡可能包括p53依赖和p53非依赖两种方式。前者E2F1的高表达可上调p14ARF表达水平,p14ARF能与MDM2结合使其失活,从而增加p53稳定性,或直接刺激p53来诱导细胞凋亡。
综上所述,在细胞衰老的类型中有增殖衰老和早熟细胞衰老,研究显示正常的细胞增殖随代次增加逐渐丢失端粒末端的序列[20],当端粒缩短到无法维持染色体结构完整性时即可以激活p14ARF,它可以抑制MDM2的功能,从而使p53稳定并激活,进而促进多种靶基因的表达,其中最重要的就是p21的表达,p21可以抑制CDK2/cyclin E复合物的活性,从而使pRb蛋白转化为非活性形式的低磷酸化或去磷酸化状态,失活的pRb与E2F转录因子结合,使得E2F不能激活细胞周期必需的基因表达,进而使细胞停滞在G0/G1期无法进入S期从而启动染色体的复制完成增殖,从而启动细胞衰老。因此增殖衰老主要依赖于p53-p21(Cip1)-pRb-E2F信号通路。
在早熟细胞衰老的研究中显示ras诱导的细胞衰老与表达量相关,低水平表达ras可导致肿瘤发生,而高水平表达ras则诱导细胞衰老。黑色素瘤可由痣细胞恶化形成,痣细胞可分化成熟,处于生长停滞或衰老状态,然而Twist表达提高会使痣细胞突破衰老状态,发展成黑色素瘤,所以提示Twist可帮助细胞突破癌基因诱发的早熟细胞衰老保护机制[21]。目前研究发现,ras诱导的细胞早熟衰老发生与p53-p21(Cip1)-pRb-E2F或p16INK4a-pRb-E2F信号通路的激活相关,p53和Rb是两个主要的细胞衰老调控因子。p53-p21(Cip1)-pRb-E2F通路激活始于ATM等对损伤DNA的探测,进而以磷酸化的形式激活p53,活化的p53可以上调p21,后者可以通过抑制CDK2/cyclin E以及CDK4/cyclin D等细胞周期调节因子的活性来抑制细胞增殖。p16INK4a是CDK4、CDK6活性的抑制剂,CDK4/6-cyclin D复合物可以使pRb磷酸化,并通过E2F的作用激活细胞周期必需基因的表达而完成细胞周期。激活的p16INK4a可以抑制CDK4/6-cyclin D复合物活性从而阻止pRb的磷酸化,从而使细胞停滞与G0/G1期无法进入S期启动染色体的复制完成细胞周期。所以细胞周期停滞是细胞衰老的一个关键特征,研究发现衰老的细胞主要含有G1期的DNA含量,因此认为衰老细胞停滞于G1期,不能顺利进入S期。
3 细胞衰老在肿瘤抑制中的作用
在前面笔者论述了有关基因在衰老和肿瘤发生之间的关系以及发生的效应通路,人们知道衰老和肿瘤是有关系的,随着年龄的增长,患肿瘤的概率会升高。这是因为当细胞衰老时,DNA损伤修复的速度赶不上DNA损伤的速度。这时,细胞可能遭受一下三种命运之一:衰老、凋亡和肿瘤。人体中大多数的细胞是先经历了衰老,然后经历不可逆地DNA损伤之后走向凋亡。在这时凋亡被认为是最后一道屏障,起着防止细胞癌变的作用。换言之,例如皮肤起皱、骨质疏松、肌肉萎缩退化、痴呆、免疫枯竭症和器官衰老,可能都是长期抑制肿瘤必须付出的代价。研究已经证实细胞衰老是机体抗肿瘤生长的重要防御机制,其中DNA损伤修复机制位于细胞抗癌的第一线,如果修复失败就可能引起细胞凋亡或细胞衰老。
已有体外研究实验表明,细胞受癌基因的刺激后可以通过发生衰老这一途径来抑制肿瘤的形成,更多的体内实验也证实了这一作用[8]。对PTEN肿瘤抑制因子失活的小鼠前列腺癌模型的研究发现PTEN缺失时,癌前病变或非致死性肿瘤中可表达衰老标志物而在恶性肿瘤却不表达,这种由癌基因诱导的细胞衰老是依赖p53的,且PTEN与p53同时缺失并不发生衰老,而是发生肿瘤[22],说明PTEN缺陷时p53参与了细胞衰老的诱导过程从而抑制了肿瘤的发生。这种结果与以前对人前列腺异常的早期阶段的研究结果是相一致的。
Suv39h1蛋白能够使组蛋白甲基化,诱导异染色质灶的形成,在衰老细胞中可以使促生长基因的异染色质沉默。研究显示ras诱导的鼠淋巴瘤衰老中发现,Suv39h1活性是必需的,Suv39h1缺陷时,则未见衰老反应。因此证实,ras诱导的细胞衰老可以抑制淋巴瘤的形成和发展,但必需Suv39h1的参与。而且研究结果显示,在Rb促进细胞衰老的过程中,也需要Suv39h1的参与,如果缺乏则Rb不能促进细胞衰老。由Suv39h1诱导的细胞衰老与淋巴瘤发生之间关系的研究也发现体内的细胞衰老是一种重要的抗肿瘤防卫机制[23]。
以上研究中,尽管所研究的组织类型不同,具体的传导途径不同,所需的癌基因也不同,但都表明由癌基因诱导的细胞衰老并不仅是细胞体外培养中所产生的现象,在体内,无论是小鼠还是在人类,确实有着抑制肿瘤发生的重要作用。癌基因诱导的细胞衰老主要发生在癌前病变中,它可以阻止癌前病变进一步向恶性肿瘤发展。与此相对的是抑癌基因,两者在癌症的生成中间相互作用,相互制约。现已发现的众多肿瘤抑制基因,其中有些具有诱导细胞分化、衰老和凋亡的作用,如果这些相关基因失活就可能使细胞摆脱衰老过程,进而引发肿瘤。
4 总结
肿瘤的发生是一个复杂精密的过程,至今还尚未完全搞清楚。肿瘤的发生也是生物进化当中一个正常的过程,每个人的体内都有可能产生肿瘤。DNA在体内复制的过程中发生突变,这是有利于物种进化的,但是在复制的过程中出现错误,也在所难免。所以生物进化是建立在基因突变的基础之上的,这就必然会带来一个严重的后果:恶性肿瘤的发生,因此如何利用细胞衰老的机制成为高危人群预防肿瘤发生的靶标是很有必要的。现在对肿瘤的治疗方法通常都是提高肿瘤抑制基因的活性,但是这种方法会不会加速患者的衰老、导致痴呆等一些与衰老有关的老年疾病?而且,一些抗衰老的药物会不会增加患癌症的风险?这些问题都有待进一步的研究探讨。
而癌基因在肿瘤防治中的作用可以诱导细胞衰老,并且这种衰老发生在癌前病变中。与恶性肿瘤不一样的是,癌前病变可停止甚至逆转。所以由癌基因诱导的衰老给肿瘤的发展设置了障碍。当机体一旦发生恶性肿瘤,肿瘤细胞中的衰老并非完全无能为力,只要肿瘤抑制基因重新表达,或者癌基因失活,肿瘤细胞仍有衰老的可能。因此细胞衰老的标记物可以作为肿瘤早期标志物,为提前发现肿瘤和肿瘤的产生提供了依据。但是在复杂的研究背后,仍然存在问题,如癌基因可以诱导衰老又可以导致癌前病变,这二者孰先孰后值得进一步的研究。
[1] d’Adda di Fagagna F.Living on a break:ellurlar senescence as a DNA-damage response〔J〕.Nat Rev Cancer,2008,8(7):512-522.
[2] Braig M,Schmitt CA.Oncogene-induced senescence:putting the brakes on tumor development〔J〕.Cancer Res,2006,66(6):2881-2884.
[3] Bringold F,Serrano M.Tumor suppressors and oncogenes in cellular senescence〔J〕.Exp Gerontol,2000,35:317-329.
[4] 何小明,梅元武.兴奋性氨基酸受体对大鼠坐骨神经切断后脊髓神经细胞的影响〔J〕.中国临床康复,2003,7(16):2306-2307.
[5] 蒋恩社,闫剑群,宋新爱.内脏不适与甜味刺激对大鼠孤束核神经元激活的相互作用〔J〕.中国临床康复,2003,7(13):1898.
[6] 王相如,钱济先,龙华,等.癌基因表达对骨肉瘤患者生存率及生活质量的意义〔J〕.中国临床康复,2003,7(8):1266-1267.
[7] Sarkisian CJ,Keister BA,Stairs DB,et al.Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis〔J〕.Nat Cell Biol,2007,9(5):493-505.
[8] Campisi J,d’Adda di Fagagna F.Cellular senescence:when bad things happen to good cells〔J〕.Nature Reviews Molecular Cell Biology,2007,8(9):729-740.
[9] Bond J,Haughton M,Blaydes J,et al.Evidence that transcriptional activation by p53plays a direct role in the induction of cellular senescence〔J〕.Oncogene,1996,13(10):2097-2104.
[10] Chin L,Artandi SE,Shen Q,et al.p53deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis〔J〕.Cell,1999,97(4):527-538.
[11] Ashcroft M,Taya Y,Vousden KH.Stress signals utilize multiple pathways to stabilize p53〔J〕.Molecular and Cellular Biology,2000,20(9):3224-3233.
[12] Carnero A,Beach DH.Absence of p21cooperates with c-myc in bypassing Ras-induced senescence and enhances oncogenic cooperation〔J〕.Oncogene,2004,23(35):6006-6011.
[13] Jarrard DF,Sarkar S,Shi Y,et al.p16/pRb pathway alterations are required for bypassing senescence in human prostate epithelial cells〔J〕.Cancer Research,1999,59(12):2957-2964.
[14] Haferkamp S,Tran SL,Becker TM,et al.The relative contributions of the p53and pRb pathways in oncogene-induced melanocyte senescence〔J〕.Aging,2009,1(6):542-556.
[15] Ye X,Zerlanko B,Zhang R,et al.Definition of pRBand p53-dependent and-independent steps in HIRA/ASF1amediated formation of senescence-associated heterochromatin foci〔J〕.Molecular and Cellular Biology,2007,27(7):2452-2465.
[16] Amati B,Alevizopoulos K,Vlach J.Myc and the cell cycle〔J〕.Frontiers in Bioscience,1998,3:d250-d268.
[17] Wang J,Xie LY,Allan S,et al.Myc activates telomerase〔J〕.Genes and Development,1998,12(12):1769-1774.
[18] Brummelkamp TR,Berns K,Hijmans EM,et al.Functional identification of cancer-relevant genes through largescale RNA interference screens in mammalian cells〔J〕.Cold Spring Harbor Symposia on Quantitative Biology,2004,69:439-445.
[19] 廖素环,施李鸣,温荣辉,等.S-腺苷高半胱氨酸水解酶在甲基循环中的代谢调节〔J〕.动物医学进展,2011,32(6):115-118.
[20] 廖亚平,汪华侨.端粒和端粒酶与细胞衰老〔J〕.国外医学:内科学分册,2005,32(9):397-400.
[21] Ansieau S,Bastid J,Doreau A,et al.Induction of EMT by twist proteins as a collateral effect of tumorpromoting inactivation of premature senescence〔J〕.Cancer Cell,2008,14:79-89
[22] Chen Z.Trotman LC,Shaffer D,et al.Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis〔J〕.Nature,2005,436(7051):725-730.
[23] Braig M,Lee S,Loddenkemper C,et al.Oncogene-induced senescence as an initial barrier in lymphoma development〔J〕.Nature,2005,436(7051):660-665.
