磷钼杂多酸催化油酸与醇酯化反应合成脂肪酸酯的研究
2012-01-13赖君玲罗根祥刘春生
赖君玲,王 婧,罗根祥,刘春生
(辽宁石油化工大学,辽宁抚顺113001)
脂肪酸酯是一种极为重要的有机化合物,在化学工业中有着广泛的应用,可以用作乳化剂、润滑剂、油品添加剂、油墨添加剂、香料、溶剂和表面活性剂等[1]。目前,由于石油资源紧缺,生物柴油成为一种最理想的可替代能源[2-4],其主要组分为脂肪酸酯。在脂肪酸酯的生产中,存在几个严重的缺点,如原材料昂贵、设备腐蚀严重、污染环境等[5-6]。酯化反应通常以浓硫酸为催化剂,这种催化剂活性高,但造成的环境污染很严重[7-10]。为了克服上述问题,研究者开发出许多固体酸催化剂,例如无机盐类[11]、离子交换树脂[12-13]、沸石分子筛[14]和杂多酸等[15-16]。杂多酸催化剂活性高,反应条件温和,易分离,能回收利用[17],是一种环境友好的催化剂。因此杂多酸催化剂被认为是有机化学反应最有效的催化剂[18-20]。本课题合成出各种磷钼酸盐催化剂,通过XRD和TG对其进行表征;考察磷钼酸盐催化剂的种类、催化剂焙烧温度、催化剂的用量、酸醇摩尔比、反应温度、反应时间对油酸与甲醇酯化反应转化率的影响,寻找最佳反应条件;考察磷钼酸锡催化剂在最佳条件下催化不同脂肪酸和不同醇的酯化反应的活性。
1 实 验
1.1 原 料
油酸、硬脂酸、月桂酸、肉豆蔻酸、甲醇、乙醇、丙醇、氢氧化钾和氯化亚锡,沈阳化学试剂厂生产;磷钼酸和硝酸铝,天津市化学试剂三厂生产;无水乙醇和95%无水乙醇,沈阳市新化试剂厂生产;氯化铋,北京顺义县李遂化工厂生产;硝酸锌,沈阳市东兴试剂厂生产;硝酸铜和硝酸铁,沈阳试剂一厂生产。以上试剂均为分析纯。
1.2 催化剂的制备和表征
在水溶液中加入4mmol的磷钼酸(HPMo),待全部溶解后,边搅拌边加入6mmol的氯化亚锡,将所得溶液放入干燥箱中于110℃干燥4h,然后在200℃下焙烧3h,焙烧后即得到催化剂磷钼酸锡(SnPMo)。磷钼酸锌(ZnPMo)、磷钼酸铜(CuPMo)、磷钼酸铝(AlPMo)、磷钼酸铁(FePMo)、磷钼酸铋(BiPMo)等磷钼酸盐(MPMo)的制备方法同上,所使用的HPMo均为4mmol,金属盐的用量分别为硝酸锌6mmol、硝酸铜6mmol、硝酸铝4mmol、硝酸铁4mmol、氯化铋4mmol。
采用日本理学公司生产的D/max-RB 12kW转靶X射线衍射仪(XRD)测定催化剂的衍射强度,测试条件:Cu Kα射线,管电压40kV,管电流100mA,采用θ-2θ连续扫描方式,步长0.02°(2θ),扫描速率为4(°)/min。
采用美国TA Instruments公司生产的SDT 2960DSC-TGA型差热-热重联用分析仪测定催化剂的热稳定性,以α-Al2O3作参比物,Ar气氛,升温速率10℃/min。
1.3 酯化反应
在100mL三口烧瓶中加入一定比例的油酸和醇,在室温下搅拌,使其充分混合后,测定反应前体系的酸值(X1);接着向三口烧瓶中加入一定量的催化剂,安装上温度计以及回流冷凝装置,磁力搅拌下开始加热,在温度(68±1)℃下反应6h,分离催化剂和反应后的混合液,测得反应结束后体系的酸值(X2)。酯化反应的转化率(y)计算如下:
y=(1-X2/X1)×100%
2 结果与讨论
2.1 催化剂的表征
不同MPMo和HPMo的XRD谱见图1。从图1可以看出,在2θ为7°~11°,16°~22°,25°~31°的范围内HPMo和MPMo都有很强的衍射峰,说明实验所制备的MPMo保持了HPMo原有的杂多酸的Keggin型结构[21],同时,金属离子取代了HPMo中的氢离子,造成了峰位置和峰强度稍有差异。
不同MPMo的热重分析结果见图2。从图2可以看出:在50~130℃范围内,不同的MPMo均有一个明显的失重峰,在80~90℃下达到最大值,分析其原因,可能是样品失去结晶水和吸附水所致;当温度升高后,ZnPMo,CuPMo,SnPMo的热重曲线变得平稳,没有明显的失重峰,表明这三种磷钼杂多酸催化剂热稳定性好;而BiPMo在220~270℃有一个明显的失重峰,FePMo在370~400℃有一个明显的失重峰,AlPMo在340~360℃有一个明显的失重峰,这可能是由于BiPMo、FePMo和AlPMo的骨架开始分解引起的,说明这三种杂多酸催化剂的热稳定性稍差。
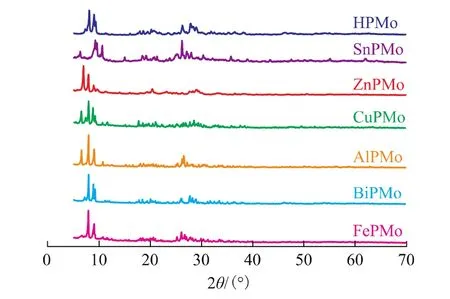
图1 HPMo和不同MPMo的XRD图谱
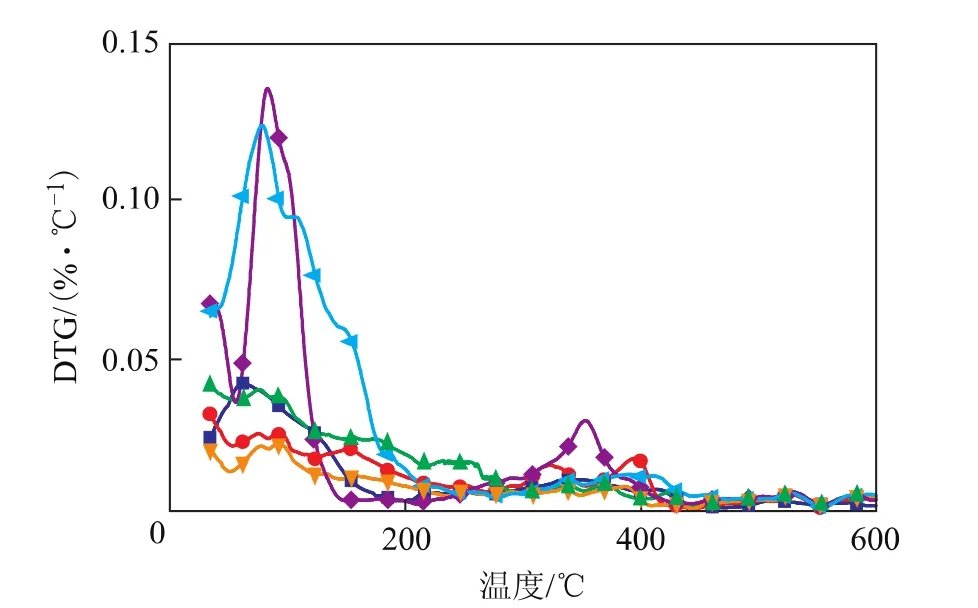
图2 不同MPMo的热重分析▲结果■—SnPMo;●—FePMo;▲—BiPMo;—CuPMo;◆—AlPMo;▲—ZnPMo
2.2 反应条件对酯化反应的影响
2.2.1 磷钼酸盐的种类对转化率的影响 在油酸用量0.075mmol、甲醇用量0.45mmol、催化剂用量为油酸质量的2%、反应温度(68±1)℃、反应时间6h的条件下,考察焙烧温度为200℃时不同MPMo对油酸和甲醇酯化反应转化率的影响,结果见表1。从表1可以看出,SnPMo的催化效果最好,转化率可达到75.3%。因此选择SnPMo为油酸与甲醇酯化反应的催化剂。
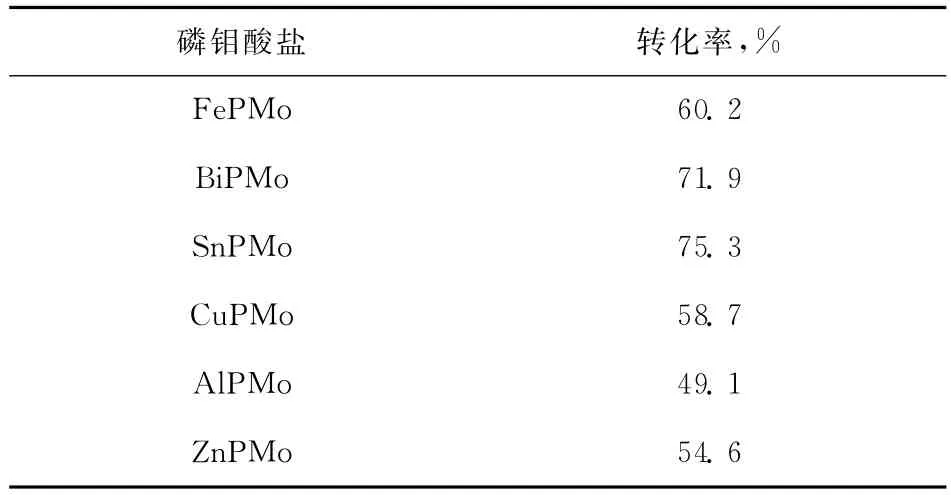
表1 MPMo对酯化反应转化率的影响
2.2.2 催化剂的焙烧温度对转化率的影响 在油酸用量0.075mmol、甲醇用量0.45mmol、催化剂SnPMo用量为油酸质量的2%、反应温度(68± 1)℃、反应时间6h的条件下,考察不同焙烧温度对SnPMo催化活性的影响,结果见表2。从表2可以看出:当催化剂SnPMo的焙烧温度由200℃升高到300℃时,转化率变化不大;当焙烧温度继续增加到400℃和500℃时,转化率大幅度下降。其原因可能是当焙烧温度升高时,SnPMo的骨架发生变化,从而造成催化剂活性下降,使酯化反应的转化率下降。
对不同焙烧温度下的SnPMo样品进行XRD表征,结果见图3。从图3可以看出:SnPMo的焙烧温度为200℃和300℃时,其骨架没有发生变化;当焙烧温度高于400℃时,其衍射峰发生了明显的变化,表明其杂多酸的骨架结构受到破坏,从而影响了其催化活性。因此SnPMo催化剂的最佳焙烧温度为200℃。
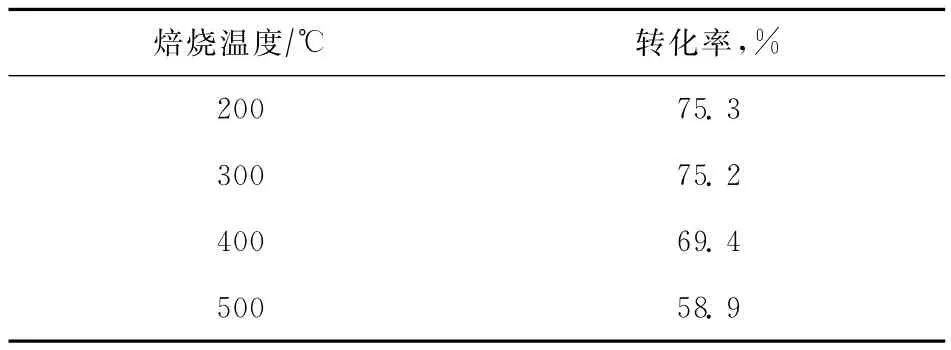
表2 SnPMo的焙烧温度对酯化反应转化率的影响
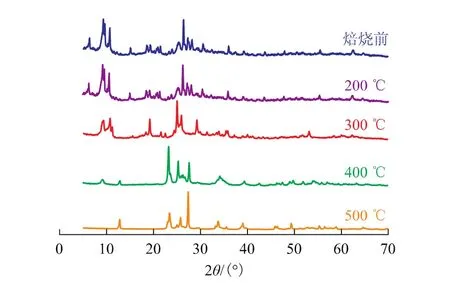
图3 不同焙烧温度下SnPMo的XRD图谱
2.2.3 酸醇摩尔比对转化率的影响 在催化剂SnPMo用量为油酸质量的2%、反应温度(68± 1)℃、反应时间6h的条件下,考察油酸与甲醇的摩尔比对转化率的影响,结果见表3。从表3可以看出:随着酸醇比的增加,转化率呈先增加后趋于稳定的变化趋势,当n(油酸)∶n(甲醇)=1∶8时,转化率最大。这是因为适量地增加甲醇用量可以使反应向酯化反应的方向进行,从而使转化率增加;而在甲醇用量达到一定程度后,反应达到平衡,继续增加甲醇用量,转化率变化不大。因此,最佳酸醇比为n(油酸)∶n(甲醇)=1∶8。

表3 酸醇摩尔比对酯化反应转化率的影响
2.2.4 催化剂用量对转化率的影响 在n(油酸)∶n(甲醇)=1∶8、反应温度(68±1)℃、反应时间6h的条件下,考察催化剂SnPMo用量对酯化反应转化率的影响,结果见图4。从图4可以看出:当催化剂用量为油酸质量的4%时,转化率达到最大值,为88.6%;继续增加催化剂的用量时,转化率变化不大。因此,适宜的催化剂SnPMo用量为油酸质量的4%。
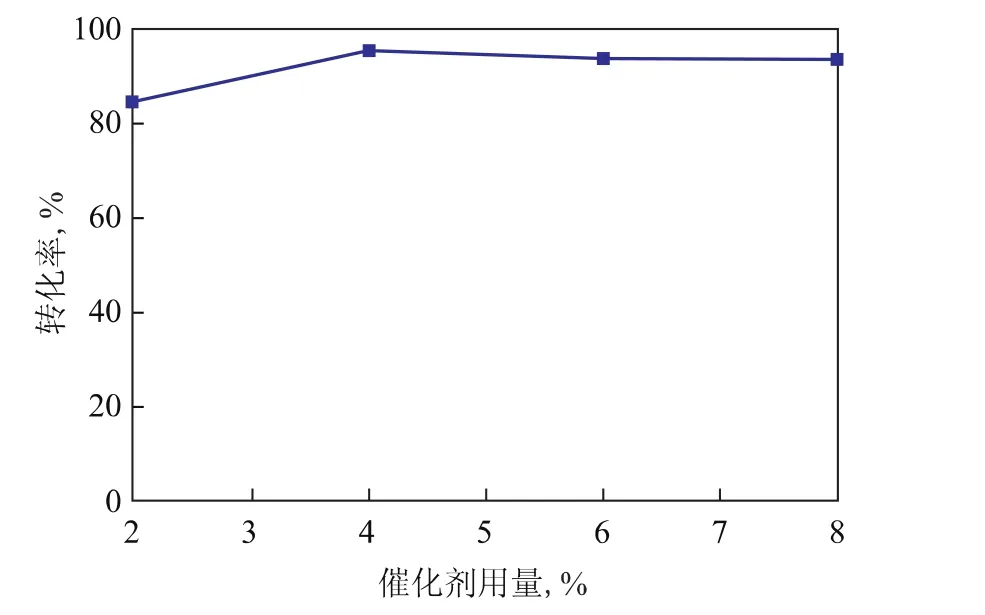
图4 催化剂用量对转化率的影响
2.2.5 反应时间对转化率的影响 在n(油酸)∶n(甲醇)=1∶8、催化剂SnPMo的用量为油酸质量的4%、反应温度(68±1)℃的条件下,考察反应时间对酯化反应转化率的影响,结果见图5。从图5可以看出:反应时间为6h时,转化率达到最大值,为95.4%;继续增加反应时间到8h时,转化率反而略微下降,这可能是因为过长的反应时间导致了一些副反应的发生。因此,适宜的反应时间为6h。
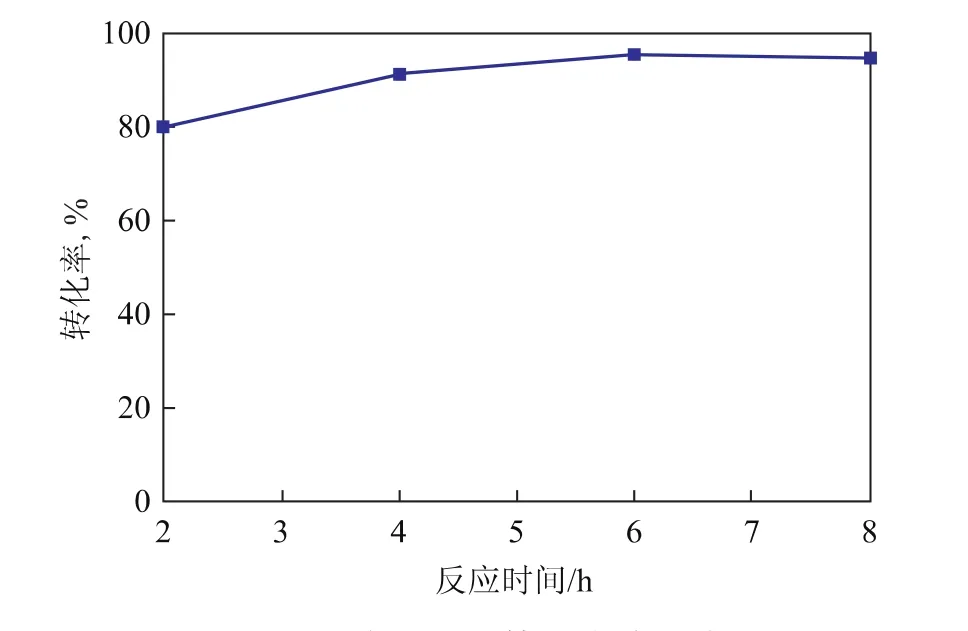
图5 反应时间对转化率的影响
2.2.6 催化剂的重复使用对转化率的影响 为了考察该催化剂的重复使用性能,待第一次反应结束后,滤出反应液,催化剂保留在圆底烧瓶中,再加入相同的反应物。在n(油酸)∶n(甲醇)=1∶8、催化剂SnPMo的用量为油酸质量的4%、反应温度(68±1)℃、反应时间6h的条件下考察催化剂的重复使用次数对转化率的影响,结果见表4。从表4可以看出,当催化剂重复使用1次时,其转化率下降了大约3百分点,当重复使用3次时,转化率下降大约10百分点,重复使用4次和5次后,转化率趋于稳定。
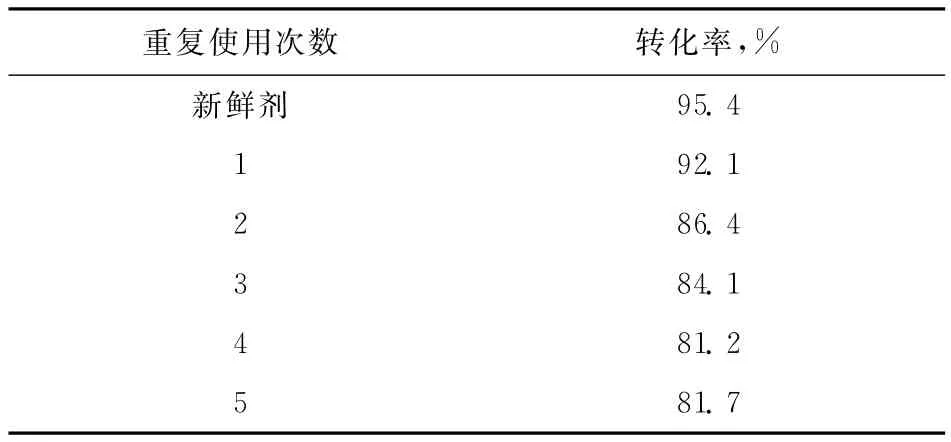
表4 催化剂重复使用次数对转化率的影响
2.3 油酸与不同醇的酯化反应
在油酸用量0.075mmol、醇用量0.6mmol、SnPMo用量为油酸质量的4%、反应温度(68± 1)℃、反应时间6h的条件下,考察SnPMo催化剂对油酸与不同低碳链醇的酯化反应活性,结果见表5。从表5可以看出,油酸和甲醇酯化反应的转化率为95.4%,而油酸和乙醇或者丙醇反应时,酯化反应的转化率分别下降为78.9%、66.3%,表明随着醇碳链长度的增加,其相应的转化率明显下降。

表5 油酸与不同醇的酯化反应转化率
2.4 不同脂肪酸与甲醇的酯化反应
在脂肪酸用量0.075mmol、甲醇用量0.6mmol、SnPMo用量为油酸质量的4%、反应温度(68± 1)℃、反应时间6h的条件下,SnPMo催化剂对不同脂肪酸与甲醇酯化反应的活性,结果见表6。从表6可以看出:不同脂肪酸与甲醇进行酯化反应的转化率在92%~97%范围内;当脂肪酸的碳链增加时,其酯化反应的转化率仅略有减小。
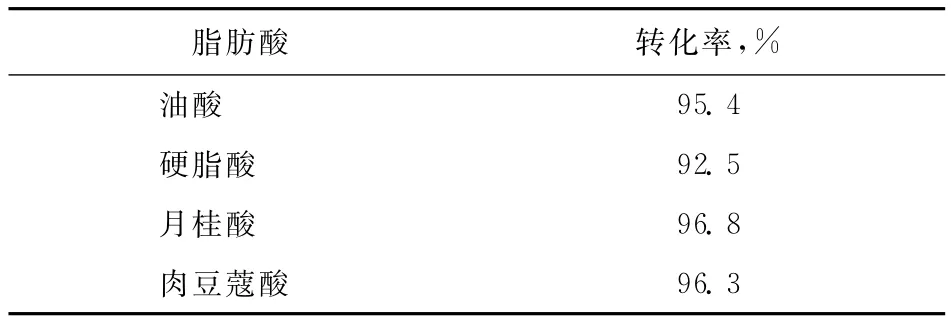
表6 不同脂肪酸与甲醇的酯化反应转化率
3 结 论
(1)以HPMo与不同金属盐反应制备的磷钼酸金属催化剂,保持了原有杂多酸的Keggin型结构,其中ZnPMo,CuPMo,SnPMo催化剂的热稳定性很好,FePMo,AlPMo和BiPMo催化剂的热稳定性稍差。
(2)SnPMo催化油酸与甲醇酯化反应的最佳条件为:n(油酸)∶n(甲醇)=1∶8、SnPMo的用量为油酸质量的4%、反应温度(68±1)℃、反应时间6h。在该条件下,油酸与甲醇酯化反应的最高转化率为95.4%。
(3)在最佳条件下,SnPMo催化不同脂肪酸与不同醇的反应活性的考察结果表明:当醇的碳链增加时,其酯化反应的转化率减小;当脂肪酸的碳链增加时,其酯化反应的转化率仅略有减小。
[1] Mantri K,Komura K,Sugi Y.ZrOCl2·8H2O catalysts for the esterification of long chain aliphatic carboxylic acids and alcohols:The enhancement of catalytic performance by supporting on ordered mesoporous silica[J].Green Chem,2005,7:677-682
[2] Serio M D,Tesser R,Pengmei L,et al.Heterogeneous catalysts for biodiesel production[J].Energy Fuel,2008,22(1):207-217
[3] Marchetti J M,Miguel V U,Errazu A F.Possible methods for biodiesel production[J].Renew Sust Energ Rev,2007,11(6):1300-1311
[4] Demirbas A.Biodiesel from bay laurel oil via compressed methanol transesterification[J].Energ Sources,Part A,2010,32(13):1158-1194
[5] Haas M J.Improving the economics of biodiesel production through the use of low value lipids as feedstocks:Vegetable oil soapstock[J].Fuel Process Technol,2005,86(10):1087-1096
[6] Bozbas K.Biodiesel as an alternative motor fuel:Production and policies in the European Union[J].Renew Sust Energ Rev,2008,12(2):542-552
[7] Altiokka M R,Citak A.Kinetics study of esterification of acetic acid with isobutanol in the presence of amberlite catalyst[J].Appl Catal A Gen,2003,239(1/2):141-148
[8] Chen Xin,Xu Zheng,Okuhara T.Liquid phase esterification of acrylic acid with 1-butanol catalyzed by solid acid catalysts[J].Appl Catal A Gen,1999,180(1/2):261-269
[9] Liu W T,Tan C S.Liquid-phase esterification of propionic acid with n-butanol[J].Ind Eng Chem Res,2001,40(15):3281-3286
[10]Peters T A,Benes N E,Holmen A,et al.Comparison of commercial solid acid catalysts for the esterification of acetic acid with butanol[J].Appl Catal A Gen,2006,297(2):182-188
[11]Konwar D,Gogoi P K,Gogoi P,et al.Esterification of car-boxylic acids by acid activated Kaolinite clay[J].Ind J Chem Technol,2008,15(1):75-78
[12]Gimenez J,Costa J,Cervera S.Vapor-phase esterification of acetic acid with ethanol catalyzed by a macroporous sulfonated styrene-divinylbenzene(20%)resin[J].Ind Eng Chem Res,1987,26(2):198-202
[13]Blagov S,Paradaa S,Bailer O,et al.Influence of ion-exchange resin catalysts on side reactions of the esterification of n-butanol with acetic acid[J].Chem Eng Sci,2006,61(2):753-765
[14]Jermy B R,Pandurangan A.A highly efficient catalyst for the esterification of acetic acid using n-butyl alcohol[J].J Mol Catal A:Chem,2005,237(1/2):146-154
[15]Verhoef M J,Kooyman P J,Peters J A,et al.A study on the stability of MCM-41-supported heteropoly acids under liquidand gas-phase esterification conditions[J].Micropor Mesopor Mater,1999,27(2/3):365-371
[16]Parida K M,Mallick S.Silicotungstic acid supported zirconia:An effective catalyst for esterification reaction[J].J Mol Catal A Chem,2007,275(1/2):77-83
[17]Juan J C,Zhang J C,Yarmo M A.Efficient esterification of fatty acids with alcohols catalyzed by Zr(SO4)2·4H2O under solvent-free condition[J].Catal Lett,2008,126(3/4):319-324
[18]Shimizu K,Niimi K,Satsuma A.Polyvalent-metal salts of heteropolyacid as efficient heterogeneous catalysts for Friedel-Crafts acylation of arenes with carboxylic acids[J].Catal Commun,2008,9(6):980-983
[19]Mouhtady O,Gaspard-Houghmane H,Roux C L.Metal triflates-methanesulfonic acid as new catalytic systems:Application to the Fries rearrangement[J].Tetrahedron Lett,2003,44(34):6379-6382
[20]Aspinall H C,Bissett J S,Greeves N,et al.Lanthanum triflate-catalysed allylation of aldehydes:Crucial activation by benzoic acid[J].Tetrahedron Lett,2002,43(2):319-321
[21]Tamon H,Okazaki M.Influence of acidic surface of activated carbon on gas adsorption characteristics[J].Carbon,1996,34(6):741-746
