4″-O-取代红霉素衍生物的研究进展
2012-01-12毕小玲
田 鑫, 毕小玲
(中国药科大学药物化学教研室,江苏南京 210009)
红霉素(erythromycin)是由美国礼来(Eli Lilly)公司于20世纪50年代开发上市的首个大环内酯类抗生素,具有疗效确切、毒性较低等优点,但其抗菌谱窄、水溶性差,且在胃酸中不稳定,易分解失活,对胃肠道有刺激作用[1-2]。
为了提高红霉素对酸的稳定性,研究人员根据红霉素的酸缩酮化失效原理,在20世纪80年代末至90年代初开发出以罗红霉素(roxithromycin)、克拉霉素(clarithromycin)、阿奇霉素(azithromycin)为代表的第2代半合成红霉素衍生物。这些衍生物虽克服了对酸的不稳定性,且药动学性质改善,抗菌谱拓宽,抗菌活性增强,胃肠道刺激等副作用明显降低,但诱发的细菌耐药现象也日趋严重[3-4]。
在研究大环内酯类抗生素的作用机制及细菌耐药机制的基础上,研究人员又开发出对耐药菌有效、抗菌活性强,且药动学性质优良的第3代红霉素类衍生物[5]。其中,酮内酯类(ketolides)抗生素取得了较大进展,已获准上市的有泰利霉素[6](telithromycin,1)和喹红霉素[7](cethromycin,ABT-773,2)。

美国辉瑞(Pfizer)公司于1998年报道的CP-544372(3)则是在克拉定糖的4″位羟基上引入氨基甲酸酯的红霉素衍生物[8],其体外抗菌活性与泰利霉素相当,对mef E基因介导的耐药肺炎链球菌的活性则约是泰利霉素的5倍,两者的MIC分别为0.08和0.39 mg·L-1;小鼠经口给药平均半衰期为6.5 h[9]。

ALLen很早就证明了红霉素C3位的克拉定糖能诱导细菌产生大环内酯类-林可霉素-链阳菌素B (MLSB)交叉耐药(Antimicrob Agent Chemother,1977年),此后有报道称在L-克拉定糖的4″位羟基上引入特定的基团会降低其对耐药性的诱导作用,对抗菌活性的影响则甚小[10]。X-射线衍射分析显示:红霉素与细菌核糖体23S rRNA结合时,L-克拉定糖处在红霉素结合位点结构域Ⅴ中由G2505、C2610和C2611组成的空腔内[11],其4″位羟基虽指向核糖体肽基转移酶中心,但尚未到达该中心,4″位上若连有氨基甲酸酯侧链则可到达该中心位置并与细菌核糖体50S亚基A~P位核苷酸靶点结合,推测将克拉定糖的4″位羟基修饰成氨基甲酸酯侧链(如化合物3)可有效克服MLSB耐药[12-14]。
基于此,近年来对克拉定糖上4″位羟基的修饰逐渐成为研究热点,其主要修饰方法是在4″位引入芳杂环取代的氨基甲酸酯、羧酸酯或醚的侧链,同时结合其他部位的结构修饰,以期获得对耐药菌有效的新型红霉素衍生物。
1 4″-氨基甲酸酯类红霉素衍生物
1.1 4″-氨基甲酸酯类化合物
Ma等[15]合成了一系列对红霉素敏感的肺炎链球菌、金葡菌和酿脓链球菌均显示出很强活性的4″-氨基甲酸酯克拉霉素衍生物,其中一些化合物对红霉素耐药型菌株也有很高的活性。如化合物4对erm B和mef A基因介导的红霉素耐药型肺炎链球菌AB11的活性是克拉霉素的512倍,两者MIC分别为0.25和128 mg·L-1;化合物5对erm B基因介导的红霉素耐药型肺炎链球菌B1的活性是克拉霉素的1 067倍,两者MIC分别为0.06和64 mg·L-1。构效关系研究表明:4″位氧原子与芳环之间相隔3~8个原子时,化合物对红霉素耐药型肺炎链球菌的活性佳。4″-O-取代侧链上的芳香基、烷基、烯基和环烷基等可能通过氢键、π-π堆积作用或范德华力增加对耐药菌核糖体的亲和力。

1.2 11,12-环碳酸酯-4″-氨基甲酸酯类化合物
Ma等[16]报道了一系列11,12-环碳酸酯-4″-氨基甲酸酯阿奇霉素衍生物。其中,化合物6和7对mef基因介导的红霉素耐药型肺炎链球菌A22072的活性(MIC=0.06 mg·L-1)均为阿奇霉素(MIC= 4 mg·L-1)的64倍,分别是未引入11,12-环碳酸酯-4″-氨基甲酸酯的阿奇霉素衍生物活性的16和8倍;化合物7对erm和mef基因介导的红霉素耐药型肺炎链球菌AB11的活性(MIC=2 mg·L-1)是阿奇霉素(MIC=256 mg·L-1)的128倍。构效关系研究表明:将15元大环内酯的4″位羟基修饰成氨基甲酸酯结构,同时使11,12位羟基形成环碳酸酯所得的阿奇霉素衍生物可有效增强对mef基因介导的红霉素耐药型肺炎链球菌的活性。

1.3 11,12-环氨基甲酸酯-4″-氨基甲酸酯类化合物
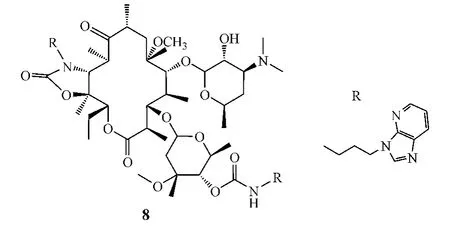
中国医学科学院药物研究所的研究人员Xu等[10]报道了一系列11,12-环氨基甲酸酯-4″-氨基甲酸酯类克拉霉素衍生物。其中,化合物8在11,12-环氨基甲酸酯和4″-氨基甲酸酯的氮原子上分别连接了2个相同的芳杂环侧链,该化合物对红霉素敏感型菌株的活性(MIC=0.5 mg·L-1)虽与阿奇霉素(MIC=0.125 mg·L-1)相当,但对红霉素耐药型肺炎链球菌03436和酿脓链球菌03740的活性(MIC分别为0.125和0.5 mg·L-1)却为克拉霉素(MIC分别为64和256 mg·L-1)和阿奇霉素(MIC分别为64和不低于256 mg·L-1)的500倍以上,且对红霉素耐药型酿脓链球菌03480的活性也强于阿奇霉素,MIC分别为0.5 mg·L-1和不低于256 mg·L-1。
1.4 4″,11-二氨基甲酸酯类化合物
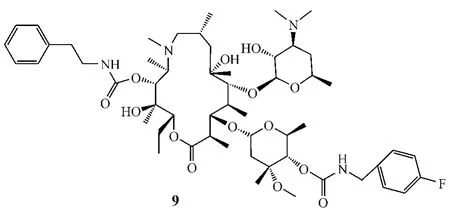
Ma等[17]合成了一系列4″,11-二氨基甲酸酯类阿奇霉素衍生物,与阿奇霉素相比,该系列化合物对红霉素耐药型菌株的活性更高。其中,化合物9对由erm介导的红霉素耐药型肺炎链球菌B1的MIC为0.25 mg·L-1,其活性是阿奇霉素(MIC=128 mg· L-1)的500倍;对由mef介导的红霉素耐药型肺炎链球菌A22072的活性(MIC=0.06 mg·L-1)是阿奇霉素(MIC=4 mg·L-1)的64倍。
2 4″-羧酸酯类红霉素衍生物
2.1 4″-喹诺酮取代的羧酸酯类化合物
葛兰素史克公司的研究小组利用拼合原理,将大环内酯骨架与喹诺酮通过适当的连接链连接得到一系列4″位支链含喹诺酮基团的大环内酯类新化学实体[18-22]。其中一些化合物对大环内酯类耐药型菌株显示出很好的活性。
例如,Fajdeti等[18]报道了一类4″-喹诺酮取代的羧酸酯类红霉素衍生物。其中,化合物10~12对外排机制介导的大环内酯耐药(M型耐药)肺炎链球菌Ci137,诱导性大环内酯耐药和内在性林可霉素、链阳霉素B耐药(iMcLSB耐药)肺炎链球菌134 GR M,以及内在性大环内酯类、林可霉素和链阳霉素B耐药(cMLSB耐药)酿脓链球菌166 GR-Micro的MIC均不高于0.125 mg·L-1,而泰利霉素对上述菌株的MIC分别为0.25、0.25和16 mg·L-1。
Škugor等[19]将大环内酯骨架与喹诺酮通过醚键延长连接链连接得到另一系列的4″-喹诺酮取代的羧酸酯类红霉素衍生物。结果显示:化合物13~15对cMLSB耐药酿脓链球菌166 GR-Micro的活性(MIC均为0.125 mg·L-1及以下)明显强于泰利霉素(MIC=16 mg·L-1);此外,化合物13对M型耐药肺炎链球菌Ci137和cMLSB耐药酿脓链球菌166 GR-Micro的活性(MIC≤0.125 mg·L-1)优于泰利霉素(MIC分别为0.25和16 mg·L-1)。在大鼠中进行的药动学实验显示:化合物14和15以溶液剂经口给药时的平均口服生物利用度分别为22%和16%;血浆清除率适中(分别占肝血流量的35%和43%),分布容积大(分别为10.4和10.5 L·kg-1),半衰期长(分别为9.4和6.7 h),适用于1日1次给药。

2.2 4″-非喹诺酮取代的羧酸酯类化合物
Hutinec等[23]合成了一系列芳(杂)环取代的羧酸酯类红霉素衍生物,其对红霉素敏感型肺炎链球菌和酿脓链球菌的活性(MIC≤0.13 mg·L-1)与阿奇霉素相当,大部分化合物对诱导性大环内酯耐药和内在性林可霉素耐药(iMcL耐药)肺炎链球菌4636的活性明显提高,是阿奇霉素的2~16倍。其中,化合物16对iMcL耐药肺炎链球菌4636和iMLSB耐药酿脓链球菌Finland 11的MIC分别为4和0.25 mg·L-1,活性分别是阿奇霉素的16倍(MIC≥64 mg·L-1)和64倍(MIC=16 mg·L-1)。

3 4″-醚类红霉素衍生物
3.1 4″-喹诺酮取代的醚类化合物
Jakopovi等[24]合成了一系列4″位支链含喹诺酮结构的4″-醚类阿奇霉素衍生物。该类化合物对红霉素敏感型革兰阳性菌(如金黄色酿脓葡萄球菌和肺炎链球菌)的活性均很强(MIC≤0.125 mg· L-1),且其中大多数化合物对cMLSB耐药酿脓链球菌166GR-Micro的活性(MIC≤0.125 mg·L-1)明显强于泰利霉素(MIC=16 mg·L-1),而化合物17和18不仅对iMcLSB耐药肺炎链球菌134 GR-M、M型耐药肺炎链球菌Ci137和cMLSB耐药酿脓链球菌166GR-Micro的活性(MIC≤0.125 mg·L-1)强于泰利霉素(MIC分别为0.25、0.5和16 mg·L-1),且对流感嗜血杆菌ATTC 49247的活性(MIC均为0.5 mg·L-1)也均高于泰利霉素(MIC=1 mg·L-1)。

3.2 11,12-环碳酸酯-4″-醚类化合物
Pavlovi等[25]合成了一系列11,12-环碳酸酯-4″-醚类阿奇霉素衍生物,其对MLSB耐药肺炎链球菌和酿脓链球菌均具有很好的活性,且对流感嗜血杆菌的活性强于泰利霉素和喹红霉素。其中,化合物19对cMLSB耐药肺炎链球菌和酿脓链球菌的MIC分别小于或等于0.125和0.5 mg·L-1,活性分别是阿奇霉素(MIC≥64 mg·L-1)的512倍和128倍,对M型耐药的金葡菌和肺炎链球菌的MIC分别是0.25和小于或等于0.125 mg·L-1,活性分别是阿奇霉素(MIC≥64 mg·L-1,MIC=4 mg·L-1)的256倍和32倍。

4 结语
目前,细菌对抗生素的耐药现象颇为严重,给临床治疗带来很大困扰,开发对耐药菌有效的第3代红霉素衍生物是当前大环内酯类抗生素研究的主要任务。其中,4″-O-取代的红霉素衍生物是近年来研究较多的一类化合物,其在对敏感菌保持良好抗菌活性的同时,对红霉素耐药型肺炎链球菌、金葡菌、流感嗜血杆菌等也有较强的抗菌活性,从而克服了第1、2代红霉素类药物会诱导细菌产生耐药性的问题。相信对该类衍生物的研究还将进一步完善人们对其构效关系的认识。
笔者所在课题组目前正致力于在克拉霉素4″位羟基上引入芳(杂)环取代氨基甲酸酯侧链的同时,对大环内酯骨架的其他部位进行修饰,以期得到对耐药菌有效的新型红霉素衍生物。初步药理活性测试结果显示其中一些衍生物对红霉素耐药型肺炎链球菌、金葡菌的活性为克拉霉素的2~32倍,相关研究将另文报道。
[1] Pal S.A journey across the sequential development of macrolides and ketolides related to erythromycin[J].Tetrahedron,2006,62(14):3171-3200.
[2] Sunitaki T,Omura S,Iwasaki S,etal.Chemicalmodification ofmacrolides in macrolide antibiotics,chemistry,biology and practice[M].Salt Lake City:Academic Press,2002:99-179.
[3] Nilius A M,Ma Z.Ketolides:the future of themacrolides?[J].Curr Opin Pharmacol,2002,2(5):493-500.
[4] Ju Y J,Xian R Q,Zhang L,et al.Synthesis and antibacterial activity of novel 4″-O-arylalkylcarbamoyl and 4″-O-((arylalkylamino)-4-oxo-butyl)carbamoyl clarithromycin derivatives[J].Bioorg Med Chem Lett,2010,20(11): 3272-3274.
[5] Ma X D,Zhang L,Wang R M,et al.Novel C-4″modified azithromycin analogs with remarkably enhanced activity against erythromycin-resistant Streptococcus pneumoniae: The synthesis and antimicrobial evaluation[J].Eur JMed Chem,2011,46(10):5196-5205.
[6] Nguyen M,Chung E P.Telithromycin:the first ketolide antimicrobial[J].Clin Ther,2005,27(8):1144-1163.
[7] Rafie S,MacDougall C,James C L.Cethromycin:a promising new ketolide antibiotic for respiratory infections[J].Pharmacotherapy,2010,30(3):290-303.
[8] Cong C,Wang H Y,Hu Y,et al.Synthesis and antibacterial activity of novel 4″-O-benzimidazolyl clarithromycin derivatives[J].Eur J Med Chem,2011,46(7): 3105-3111.
[9] Wu Y J,Su W G.Recent developments on ketolides and macrolides[J].Curr Med Chem,2001,8(14): 1727-1758.
[10]Xu P,Liu L,Jin Z P,et al.Synthesis and antibacterial activity of 4″-O-heteroarylcarbamoyl derivatives ofmacrolide[J].Bioorg Med Chem Lett,2008,18(20): 5507-5511.
[11]Asaka T,Manaka A,Sugiyama H.Recent developments in macrolide antimicrobial research[J].Curr Top Med Chem,2003,3(9):961-989.
[12] Shen X C,Jiao B,Ma S T.Synthesis and antibacterial activity of 4″-O-carbamoyl analogs of clarithromycin[J].Chin Chem Lett,2010,21(3):257-260.
[13]Takashima H.Structural consideration of macrolide antibiotics in relation to the ribosomal interaction and drug design[J].Curr Top Med Chem,2003,3(9):991-999.
[14] Blanchard S C,Cooperman B S,Wilson D N.Probing translation with small-molecule inhibitors[J].Chem Biol,2010,17(6):633-645.
[15]Ma S T,Jiao B,Ju Y J,et al.Synthesis and antibacterial evaluation of novel clarithromycin derivatives with C-4″elongated arylalkyl groups against macrolide-resistant strains[J].Eur JMed Chem,2011,46(2):556-566.
[16]Ma ST,Ma R X,Liu Z P,et al.Synthesis and antibacterial activity of novel 15-membered macrolide derivatives: 4″-Carbamate,11,12-cyclic carbonate-4″-carbamate and 11,4″-di-O-arylcarbamoyl analogs of azithromycin[J].Eur JMed Chem,2009,44(10):4010-4020.
[17]Ma ST,Jiao B,Liu Z P,et al.Synthesis and antibacterial activity of 4″,11-di-O-arylalkylcarbamoyl azithromycin derivatives[J].Bioorg Med Chem Lett,2009,19(6): 1698-1701.
[18]Fajdeti A,Paljetak H ˇC,Lazarevski G,et al.4″-O-(ω-Quinolylamino-alkylamino) propionyl derivatives of selected macrolideswith the activity against the key erythromycin resistant respiratory pathogens[J].Bioorg Med Chem,2010,18(17):6559-6568.
[19]Škugor M M,Štimac V,Palej I,et al.Synthesis and biological activity of 4″-O-acyl derivatives of14-and 15-membered macrolides linked toω-quinolone-carboxylic unit[J].Bioorg Med Chem Lett,2010,18(17):6547-6558.
[20]Kapi S,Paljetak HˇC,Alihodži S,et al.6-Alkylquinolone-3-carboxylic acid tethered to macrolides synthesis and antimicrobial profile[J].Bioorg Med Chem Lett,2010,18 (17):6569-6577.
[21]Kapi S,Paljetak H ˇC,Jakopovi I P,et al.Synthesis of macrolones with central piperazine ring in the linker and its influence on antibacterial activity[J].Bioorg Med Chem Lett,2011,19(23):7281-7298.
[22]Štimac V,Škugor M M,Jakopovi IP,et al.Initial scaleup and process improvements for the preparation of a lead antibacterial macrolone compound[J].Org Process Res Dev,2010,14(6):1393-1401.
[23]Hutinec A,Derek M,Lazarevski G,et al.Novel 8a-aza-8a-homoerythromycin-4″-(3-substituted-amino)propionates with broad spectrum antibacterial activity[J].Bioorg Med Chem Lett,2010,20(11):3244-3249.
[24]Jakopovi IP,Kragol G,Forrest A K,et al.Synthesis and properties ofmacrolones characterized by two ether bonds in the linker[J].Bioorg Med Chem Lett,2010,18(17): 6578-6588.
[25]Pavlovi D,Mutak S.Discovery of 4″-ether linked azithromycin-quinolone hybrid series:influence of the central linker on the antibacterial activity[J].Med Chem Lett,2011,2(5):331-336.
