鬼臼毒素衍生物的构效关系分析及相关新药的研发思路
2012-01-12张仕金徐云根
张仕金, 朱 雄, 徐云根*
(1.中国药科大学药物化学教研室,江苏 南京210009;2.中国药科大学医药化工研究所,江苏南京210009)
鬼臼毒素(podophyllotoxin,1)来源于盾叶鬼臼(podophyllum)的根茎提取物,早在19世纪初就被用于催吐、导泻和驱虫。研究表明:盾叶鬼臼具有抗有丝分裂活性,鬼臼毒素便是其抗有丝分裂的活性成分(Imbert,Biochimie,1998年),且其抗有丝分裂机制与秋水仙碱类似,即通过与微管蛋白结合进而阻止微管蛋白聚合形成微管。然而,鬼臼毒素的水溶性差,生物利用度低,限制了其作为抗肿瘤药物的应用。为了提高其生物利用度,山德士公司的研究人员在对强心苷化合物洋地黄和糖苷配基化合物毒毛旋花子的研究基础上,确立了开发鬼臼毒素糖苷衍生物的研究思路,合成了一系列鬼臼毒素糖苷衍生物。其中,依托泊苷(etoposide,2)、替尼泊苷(teniposide,3)和依托泊苷磷酸盐(etopophos,4)已在临床上用于治疗小细胞肺癌、白血病、乳腺癌等,并显现出良好的疗效,而依托泊苷更已成为肿瘤的一线治疗药物,常与其他抗肿瘤药物联合治疗各种癌症,如与贝伐单抗联用于恶性胶质瘤,与氯法拉滨和环磷酰胺联用于血液系统恶性肿瘤,或与长春新碱、阿霉素和地塞米松联合治疗卡波西肉瘤[1-3]。与依托泊苷相比,依托泊苷磷酸盐临床疗效更持久,水溶性更佳。由于糖基侧链的引入,依托泊苷的作用机制发生变化,其主要通过与拓扑异构酶Ⅱ结合,进而阻止了完整DNA链的形成,在细胞分裂S期和G2期阻止细胞的形成,从而发挥抗肿瘤作用。很多研究都表明依托泊苷是通过形成DNA-药物-拓扑异构酶Ⅱ复合物,最终抑制 DNA配对从而产生DNA单链及双链片段,使肿瘤细胞周期终止于G期[4]。但是,这些鬼臼毒素糖苷衍生物仍存在骨髓抑制、中性粒细胞减少症和恶心呕吐等毒副作用。为此,研究人员选择分别在鬼臼毒素A、C、D和E环上进行结构修饰,以期获得抗肿瘤活性更佳、毒副作用更小的鬼臼毒素类化合物。

1 A环的修饰
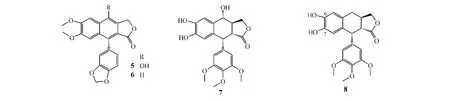
天然产物中,与鬼臼毒素结构相似的化合物有山荷叶素(diphyllin,5)、爵床脂素B(justicidin B,6)和希基鬼臼毒素(sikkimotoxin,7),这些化合物与鬼臼毒素的主要不同之处在于前者无A环,此处为开环结构,而后者此处有一亚甲基二氧环结构。有研究表明:希基鬼臼毒素及其衍生物具有细胞毒性并能在细胞有丝分裂中阻止纺锤体的形成(Stahelin,Prog Drug Res,1989年),因此,科研人员对鬼臼毒素结构中的A环进行了结构修饰。例如,Wang等(JMed Chem,1992年)合成了一系列6,7-二羟基-4β取代鬼臼毒素类似物。然而,体外活性测试结果显示:与相应的保持6,7-位为亚甲基二氧环结构的化合物相比,该系列化合物对拓扑异构酶Ⅱ的抑制活性大大降低。Castro等[5]在其合成的一系列A环衍生物中也发现了类似现象:6,7-位为羟基的衍生物(8)对白血病P-388细胞、肺癌A-549细胞、结肠癌HT-29细胞的活性明显低于化合物9(两者的IC50分别为1.3 vs0.003,1.3 vs 0.005,1.3 vs 0.005μmol·L-1)。Cho等(JMed Chem,1996年)合成了一系列A环吩嗪衍生物,并用人口腔癌KB细胞进行了细胞毒活性测试,结果发现,A环修饰后的化合物,如化合物10的活性[IC50=(0.11±0.03)μmol·L-1]明显弱于A环未作修饰的化合物(11)[IC50=(0.032± 0.008)μmol·L-1],由此认为随着A环的改变,化合物的活性下降,这进一步印证了A环保持完整的重要性。

2 C环的修饰
目前,研究人员对鬼臼毒素的结构改造主要集中在C环,进入临床应用的抗肿瘤药物依托泊苷、替尼泊苷和依托泊苷磷酸盐便是此类衍生物。另有几个很有潜力的候选药物也是经C环修饰的鬼臼毒素衍生物,如:处于Ⅱ期临床研究中的NK-611 (12)、GL-331(13)和处于Ⅰ期临床研究中的TOP-53(14)。其中,NK-611为依托泊苷的类似物,其因在C环的取代基上了引入了二甲氨基,使分子的成盐性增加,生物利用度得以提高[6]。GL-331则是通过将依托泊苷糖基团替换为非糖基团而得到的化合物,药理研究结果显示其亦可通过抑制拓扑异构酶Ⅱ活性发挥抗肿瘤作用[7]。Taiho公司研发的TOP-53对非小细胞肺癌的抑制活性为依托泊苷的10倍,在治疗转移性肺癌方面疗效良好[8-9]。

对C环的修饰通常选择的是4位上的取代基,现已分别合成了一系列4位含氧衍生物和4位含氮衍生物,并初步考察了这些化合物的抗肿瘤活性。
2.1 4位含氧衍生物 研究人员对鬼臼毒素(或4'-去甲基鬼臼毒素)4位上羟基进行修饰后得到4位含氧衍生物。例如,Duca等[10]合成了一系列4β-氧衍生物,其中化合物15对白血病L1210细胞的抑制活性为依托泊苷的20倍,进一步的研究表明其对拓扑异构酶Ⅱ也具有抑制活性。Kamal等[11]合成了一系列4β-氧甲酰氨基衍生物,其中化合物16对乳腺癌、肺癌、卵巢癌等癌细胞的抑制活性均强于依托泊苷(见表1)。Xiang等[12]利用拼合原理,将5-氟尿嘧啶(5-FU)与4'-去甲基鬼臼毒素拼合得化合物17,活性测定结果显示:该化合物在10μmol· L-1浓度下对白血病P-388细胞、肺癌A-549细胞和肝癌Bel-7402细胞的抑制率均大于依托泊苷和5-FU(见表2)。Passarella等[13]将鬼臼毒素的4位通过连接臂与硫代秋水仙碱拼合得化合物18。将鬼臼毒素和化合物18的溶液(10μmol·L-1)分别与纯化后的牛微管蛋白(3 g·L-1)混合并于37℃下孵育15 min,加入 GTP诱导微管蛋白聚合,30 min后离心分离聚合和未聚合微管蛋白,并采用光密度测定法定量分析。结果显示:在化合物18存在的条件下,聚合微管蛋白与未聚合微管蛋白的比例为(1.36±0.29);而在鬼臼毒素存在的条件下该比例为(0.80±0.1),表明化合物18抑制微管蛋白聚合的能力弱于鬼臼毒素,提示并非所有化合物均可通过拼合原理连接不同抗肿瘤药物以达到协同作用。此外,Yu等[14]合成的一系列4β-氧衍生物中也未出现理想中活性更好的化合物:在用宫颈癌HeLa细胞、卵巢癌 SKOV3细胞、白血病 K562和K562ADR细胞进行的活性测试中,仅化合物19的活性总体上与鬼臼毒素相当[两者的IC50分别为(0.55±0.06)vs(0.10±0.02),(0.31±0.04)vs (0.17±0.02),(0.03±0.01)vs(0.03±0.01),(0.30±0.05)vs(0.17±0.02)μmol·L-1]。


表1 化合物16和依托泊苷抗肿瘤活性比较Table1 Comparison of anti-tumor activity data of compound 16 and etoposide

表2 化合物17在浓度为10μmol·L-1时对肿瘤细胞的抑制率Table2 Inhibitory rates against tumor cells at 10μmol·L-1 of compound 17
2.2 4位含氮衍生物 Reddy和Bhat等人在鬼臼毒素(或4'-去甲基鬼臼毒素)的4位引入三氮唑基团,得一系列4位氮衍生物,体外活性测试结果显示:化合物20、21对胶质瘤SF-295细胞、前列腺癌PC-3细胞和喉癌Hep-2细胞的抑制活性基本上优于依托泊苷(见表3)[15-16]。Zhang等[17]合成了一系列4位5-FU取代的衍生物,肿瘤细胞活性测试结果显示该类化合物均具有较强的细胞毒性,且5-FU的引入使鬼臼毒素类化合物的水溶性得到改善,有利于生物利用度的提高,化合物22为其代表性化合物。Duca等[18]合成了一系列具有拓扑异构酶Ⅱ抑制活性的4β-氨基甲酸酯衍生物,其中化合物23的抑制活性与依托泊苷相当(IC50=20μmol·L-1),但细胞毒性(IC50=0.18μmol·L-1)强于依托泊苷(IC50=0.83μmol·L-1)。体外活性研究还显示:随着酯侧链长度的延长,氨基甲酸酯衍生物对白血病细胞L2010的抑制活性下降。而Xi等[19]和Wang等[20]对各自合成的系列4β-氨基芳环衍生物进行的体外活性测试结果显示:化合物24对肺癌A-549细胞和乳腺癌 MCF-7细胞的 IC50分别为0.71和0.92μmol·L-1,优于GL-331(IC50分别为3.54和2.13μmol·L-1);化合物25对口腔癌KB细胞的 IC50为 1.91μmol·L-1,强于依托泊苷(IC50=4.61μmol·L-1),且水溶性更好,更有利于口服吸收。此外,Kamal等[21-23]从 Becker等人(Bioorg Med Chem Lett,1998年)的研究中得知多芳环基团可增强化合物抗肿瘤活性,因此在鬼臼毒素的4位引入多芳环的疏水性基团,合成了一系列4β-氨基多芳环衍生物,其中大多数化合物对结肠癌细胞、肺癌细胞和肝癌细胞等均显示出较强的细胞毒性,但非极性大基团的引入,也导致了生物利用度的下降。
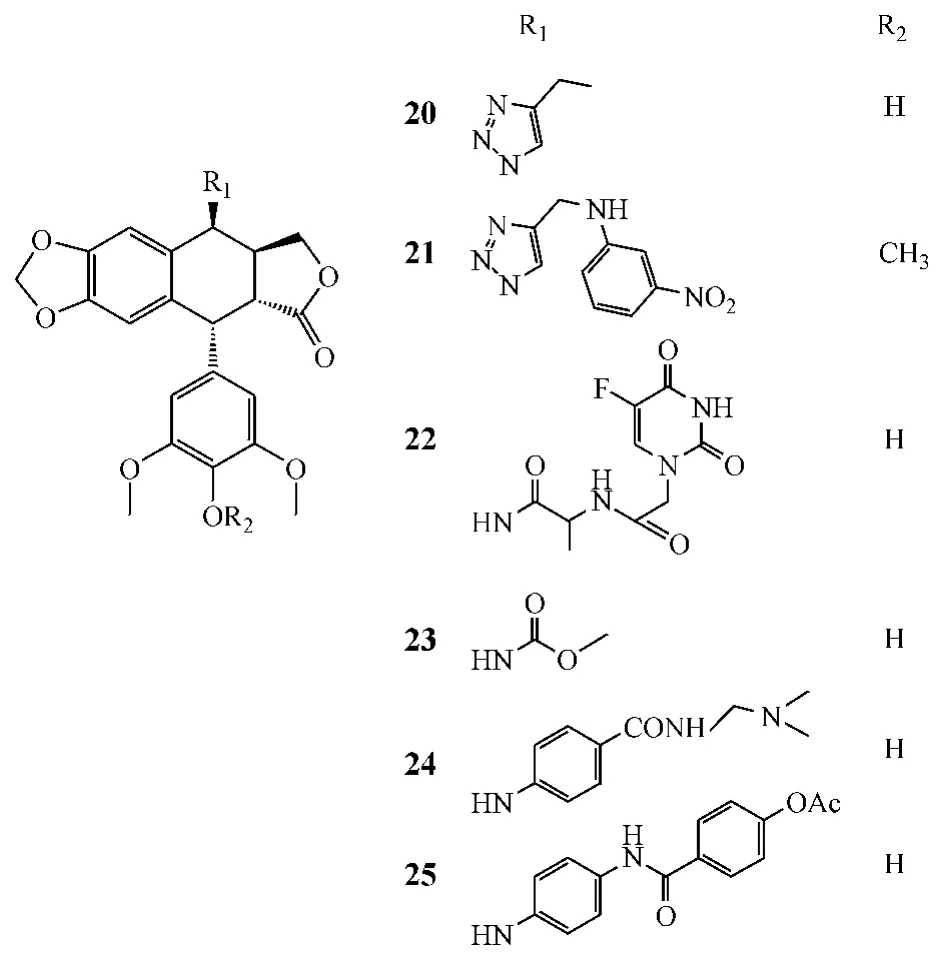
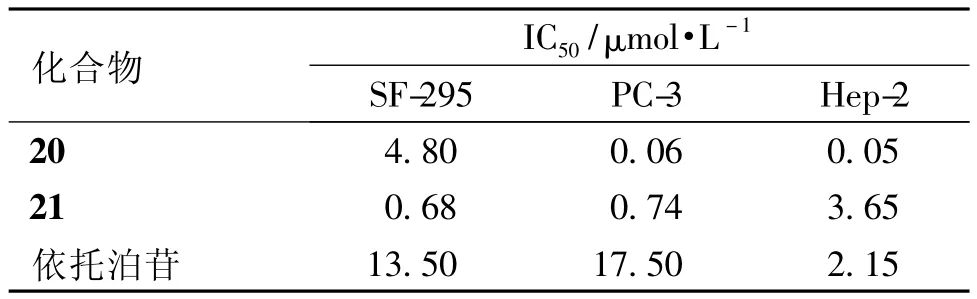
表3 化合物20、21和依托泊苷抗肿瘤活性比较Table3 Comparison of anti-tumor activity data of compound 20,21 and etoposide
3 D环和E环的修饰
目前对鬼臼毒素D环和E环的研究相对较少,Castro等[24]通过打开D环,合成了一系列细胞毒性较强的衍生物,其活性浓度均低至微摩尔级。该研究还发现当9位为醛基或亚胺基,9'位为酯基时,活性保持得最好,如化合物26。
至于E环的改造,因通过对依托泊苷的结构研究已发现4'位为羟基时活性最好,故在结构改造时通常是先在4'位羟基上连接能被体内酶水解脱落的基团,即做成前药,在体内重新释放出羟基生成原形药物,以增加吸收,提高药效。此外,在峨参的提取物中分离出的鬼臼毒素类似物——去氧鬼臼毒素(deoxypodophyllotoxin,27)已被证明具有良好的抗肿瘤活性[25]。科研人员通过在该化合物的4'位上引入一些极性基团以增加水溶性,提高生物利用度。例如,Chen等[26]在化合物27的4'位引入氨基酸基团,所得化合物的脂水分配系数的对数值(log P值)大多低于2,水溶性大于其前体化合物去氧鬼臼毒素(log P=3.16),且体外细胞毒活性测试结果显示:这些化合物对白血病细胞HL-60的IC50大多低于1μmol·L-1,优于依托泊苷(IC50=2.85μmol·L-1),对宫颈癌SiHa细胞和肺癌A549细胞的抑制活性(IC50均值分别为3.13和2.87μmol·L-1)明显强于依托泊苷(IC50分别为40.4和24.9μmol·L-1),而对宫颈癌Hela细胞的抑制活性与依托泊苷相当。Jin等[27]则在化合物27的4'位引入哌啶衍生物得化合物28,其哌啶环可形成季铵盐,从而有望增加化合物的水溶性。体外活性测定结果显示:其对肺癌A-549细胞、宫颈癌Hela和 SiHa细胞的 IC50分别为0.102、0.18、0.019 5μmol·L-1;均强于依托泊苷(IC50分别为14.8、50.6和30.7μmol·L-1)。

4 结语
综上所述,鬼臼毒素衍生物的结构改造及构效关系有如下特点:
1 )A环亚甲基二氧环的结构完整对保持活性很重要(Wang等,JMed Chem,1992年)。
2 )C环4位是结构改造的主要部位,与4位为α构型的化合物相比,4位为β构型的化合物活性更强[14],在该位置可通过-N-和-O-引入糖配体或其他非糖结构。
3)D环与C环为反式稠合,D环打开后所得化合物仍具有一定的活性[24]。
4 )E环为α构型,结构改造主要集中在4'位,该位置为-OH时方显示出良好活性,故可利用前药原理,在羟基上引入极性基团以达到增溶效果,引入的基团在体内可通过酶或非酶作用脱去。
依托泊苷水溶性差,故临床上常通过加入助溶剂的方式改善依托泊苷的溶解性,但助溶剂的加入可能会带来意想不到的副作用。尽管依托泊苷磷酸盐的研发改善了依托泊苷的水溶性问题,但随着该类药物的推广使用,也已出现了耐药性的问题,因此,进一步开发毒性小、疗效高的鬼臼毒素新型衍生物,克服现有药物的耐药性,已成为抗肿瘤药物研究的热点。
目前进行的研究工作主要集中在鬼臼毒素的C4位以及去氧鬼臼毒素的C4'位取代基上,根据生物电子等排、拼合和前药原理等进行新药设计,一些具有较强抗肿瘤活性的新化合物被相继发现。笔者所在课题组正在以去氧鬼臼毒素为先导物进行结构修饰,已取得一些阶段性结果。随着对DNA拓扑异构酶Ⅱ的作用机制的阐明,基于该酶结构进行的鬼臼毒素类化合物的结构改造将有可能帮助我们发现更安全和更高效的抗肿瘤候选化合物。
[1] Fu B D,Linskey M E,Bota D A.Bevacizumab and etoposide combination chemotherapy in patients with recurrent malignantgliomaswho failed bevacizumab[J].Drugs Ther Stud,2012,2(1):26-28.
[2] Inaba H,Bhojwani D,Pauley J L,et al.Combination chemotherapy with clofarabine,cyclophosphamide,and etoposide in children with refractory or relapsed haematologicalmalignancies[J].Br J Haematol,2012,156(2): 275-279.
[3] Zhong D T,ShiCM,Chen Q,etal.Etoposide,vincristine,doxorubicin and dexamethasone(EVAD)combination chemotherapy as second-line treatment for advanced AIDS-related Kaposi's sarcoma[J].J Cancer Res Clin Oncol,2012,138(3):425-430.
[4] Srivastava V,Negi A S,Kumaar JK,et al.Plant-based anticancermolecules:a chemical and biological profile of some important leads[J].Bioorg Med Chem,2005,13 (21):5892-5908.
[5] Castro A,Miguel del Corral JM,Gordaliza M,et al.Synthesis and cytotoxicity of podophyllotoxin analoguesmodified in the A ring[J].Eur JMed Chem,2003,38(1):65-74.
[6] You Y.Podophyllotoxin derivatives:current synthetic approaches for new anticancer agents[J].Curr Pharm Des,2005,11(13):1695-1717.
[7] Kamal A,Suresh P,Ramaiah M J,et al.Synthesis and biological evaluation of 4β-sulphonamido and 4β-[(4'-sulphonamido)benzamide]podophyllotoxins as DNA topoisomerase-IIαand apoptosis inducing agents[J].Bioorg Med Chem,2012,20(6):2054-2066.
[8] Gentry A C,Pitts S L,Jablonsky M J,et al.Interactions between the etoposide derivative F14512 and human type II topoisomerases:implications for the C4 sperminemoiety in promoting enzyme-mediated DNA cleavage[J].Biochemistry,2011,50(15):3240-3249.
[9] Byl JAW,Cline SD,Utsugi T,etal.DNA topoisomerase IIas the target for the anticancer drug TOP-53:mechanistic basis for drug action[J].Biochemistry,2001,40(3): 712-718.
[10]Duca M,Guianvarc'h D,Meresse P,et al.Synthesis and biological study of a new series of 4'-demethylepipodophyllotoxin derivatives[J].J Med Chem,2005,48(2): 593-603.
[11]Kamal A,Kumar B A,Suresh P,etal.Synthesis of4β-carbamoyl epipodophyllotoxins as potential antitumor agents[J].Bioorg Med Chem,2011,19(9):2975-2979.
[12]Xiang R,Zhang FM,Ni JM,et al.Synthesis and stability evaluation of 5-FU-acetic podophyllic ester as anti-tumor agent[J].JChin Pharma Sci,2007,16(1):47-50.
[13]Passarella D,Peretto B,Yepes R B,et al.Synthesis and biological evaluation of novel thiocolchicine-podophyllotoxin conjugates[J].Eur J Med Chem,2010,45(1): 219-226.
[14]Yu P F,Chen H,Wang J,et al.Design,synthesis and cytotoxicity of novel podophyllotoxin derivatives[J].Chem Pharm Bull,2008,56(6):831-834.
[15]Reddy D M,Jada S,Gousia C,et al.4β-[(4-Alkyl)-1,2,3-triazol-1-yl] podophyllotoxins as anticancer compounds:Design,synthesis and biological evaluation[J].Eur JMed Chem,2011,46(6):1983-1991.
[16]Bhat B A,Reddy PB,Agrawal SK,etal.Studies on novel 4β-[(4-substituted)-1,2,3-triazol-1-yl] podophyllotoxins as potential ancancer agents[J].Eur JMed Chem,2008,43(10):2067-2072.
[17]Zhang FM,Yao X J,Tian X,et al.Synthesis and biological evaluation of new 4β-5-Fu-substituted 4'-demethylepipodophyllotoxin derivatives[J].Molecules,2006,11(11): 849-857.
[18]Duca M,Arimondo P B,Léonce S,et al.Novel carbamate derivatives of 4-β-amino-4'-O-demethyl-4-desoxypodophyllotoxin as inhibitors of topoisomerase II:synthesis and biological evaluation[J].Org Biomol Chem,2005,3(6): 1074-1080.
[19]XiW L,Cai Q,Tang Y B,et al.Design and synthesis of novel cytotoxic podophyllotoxin derivatives[J].Chin Chem Lett,2010,21(10):1153-1156.
[20]Wang L,Yang F Y,Yang X C,et al.Synthesis and biological evaluation of 4β-anilino-4'-O-demethyl-4-desoxypodophyllotoxin derivatives as potential antitumor agents[J].Eur JMed Chem,2011,46(1):285-296.
[21]Kamal A,Kumar B A,Suresh P,et al.Synthesis of 4β-N-polyaromatic substituted podophyllotoxins:DNA topoisomerase inhibition,anticancer and apoptosis-inducing activities[J].Bioorg Med Chem,2010,18(24): 8493-8500.
[22]Kamal A,Kumar B A,Suresh P,etal.An efficientone-pot synthesis of benzothiazolo-4β-anilino-podophyllotoxin congeners:DNA topoisomerase-II inhibition and anticanceractivity[J].Bioorg Med Chem Lett,2011,21(1): 350-353.
[23]Kamal A,Gayatri N L,Reddy D R,et al.Synthesis and biological evaluation of new 4β-anilino-and 4β-imido-substituted podophyllotoxin congerners[J].Bioorg Med Chem,2005,13(22):6218-6225.
[24]Castro M A,Miguel del Corral JM,Garcia PA,etal.Synthesis and biological evaluation of new podophyllic aldehyde derivatives with cytotoxic and apoptosis-inducing activities[J].JMed Chem,2010,53(3):983-993.
[25]Muto N,Tomokuni T,Haramoto M,etal.Isolation ofapoptosis-and differentiation-inducing substances toward human promyelocytic leukemia HL-60 cells from leaves of Juniperus taxifolia[J].Biosci Biotechnol Biochem,2008,72(2):477-484.
[26]Chen SW,Gao Y Y,Zhou N N,et al.Carbamates of 4'-demethyl-4-deoxypodophyllotoxin:Synthesis,cytotoxicity and cell cycle effects[J].Bioorg Med Chem Lett,2011,21 (24):7355-7358.
[27]Jin Y,Liu J,Huang W T,et al.Synthesis and biological evaluation of derivatives of 4-deoxypodophyllotoxin as antitumor agents[J].Eur J Med Chem,2011,46(9): 4056-4061.
